Глава 14 гидрогенизационные процессы в нефтепереработке
Глава 14
ГИДРОГЕНИЗАЦИОННЫЕ ПРОЦЕССЫ В НЕФТЕПЕРЕРАБОТКЕ
14.1. КЛАССИФИКАЦИЯ ПРОЦЕССОВ
Гидрогенизационные процессы нашли широкое применение в нефтепереработке и нефтехимии. Их используют для получения стабильных высокооктановых бензинов, улучшения качества дизельных и котельных топлив, а также смазочных масел. В нефтехимической промышленности с помощью реакций гидрирования получают циклогексан и его производные, многие амины, спирты и ряд других мономеров.
Таблица 14.1. Доля процессов гидроочистки в различных странах мира (в % от прямой перегонки)
| Процесс |
СНГ | США |
Англия | Италия |
Франция | Япония |
ФРГ |
| Г кдроочнстка сырья для каталитического ри-формиига | 6,2 | 19,4 |
14,5 | 8,8 | 11,4 |
9,0 | 11,6 |
|
Гидроочистка средних дистиллятов |
19,2 | 31,2 | 20,6 | 10,6 | 16,1 |
15,2 | 17,3 |
| Г идрообессеривание остатков | 10,3 | 13,3 | 10,8 | 14,7 | 37,5 |
20,9 |
Быстрое развитие гидрогенизационных процессов в последние годы объясняется повышением требований к качеству товарных нефтепродуктов, значительным снижением стоимости производства водорода и созданием высокоэффективных катализаторов.
В нефтеперерабатывающей промышленности гидрогенизационные процессы используют для регулирования углеводородного и фракционного состава перерабатываемых нефтяных фракций, удаления из них серо- и азотсодержащих соединений» улучшения эксплуатационных характеристик нефтяных топлив* масел и сырья для нефтехимии.
Основные гидрогенизационные процессы следующие:
1) гидроочистка нефтяных фракций от серо-, азот- и кисло-родорганических соединений с целью повышения качества продуктов или подготовки к дальнейшей переработке;
2) гидрирование алкенов и аренов в нефтяных фракциях;
3) гидрокрекинг нефтяных фракций.
По масштабам переработки ведущее место занимают процессы гидроочистки (табл. 14.1).
14.2. ГИДРООЧИСТКА
Гидроочистка — процесс удаления из нефтепродуктов гете-роатомных, непредельных соединений и частично полицикли-ческих аренов в среде водорода на катализаторах.
Химические основы процесса. Удаление гетероатомов происходит в результате разрыва связей С—S, С—N и С—О и насыщения образующихся осколков водородом. При этом сера, азот и кислород выделяются соответственно в виде H2S, NH3 и Н20. Алкены присоединяют водород по двойной связи. Частично гидрируются полициклические арены.
Превращения серосодержащих соединений. Меркаптаны превращаются в углеводород и сероводород:
Сульфиды гидрируются через стадию образования меркаптанов:
RSR' R'SH —V R'H + H2S.
RH
Дисульфиды гидрируются до сероводорода и соответствующих углеводородов также через стадию образования меркаптанов:
RSSR' RSH+R'SH RH+R'H + 2H2S.
В циклических сульфидах, например тиофане, вначале разрывается кольцо, затем отщепляется сероводород и образуется соответствующий углеводород:
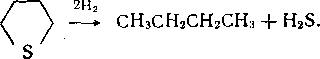
Тиофен, бенз- и дибензтиофен сначала гидрируются до производных тетрагидротиофена, которые затем превращаются в алканы и алкилпроизводные аренов:
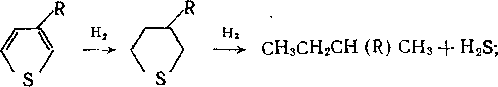
С2Н5
R
R
R
+ H2S.
С повышением температуры (в интервале 20—500 °С) константа равновесия гидрирования меркаптанов, сульфидов и дисульфидов возрастает, а тетрагидротиофенов и тиофенов — падает. Поэтому глубокая очистка нефтепродуктов от серы, содержащейся в виде тиофенов, возможна только при относительно низкой температуре (^425°С) и высоком парциальном давлении водорода (3 МПа и выше).
Кинетика гидрирования сернистых соединений сильно зависит от их строения. Скорость реакции падает в следующем ряду (в скобках — относительные скорости гидрирования): меркаптаны (7) =дибсн:шлсульфиды (7) >ото/?-алкилсульфиды (4,3— 4,4) > тиофан и его производные (3,8—4,1 )> первичные алкил-сульфиды (3,2) > производные тиофена и диарилсульфиды
В пределах одного класса соединений скорость гидрирования уменьшается с увеличением молекулярной массы, т. е. удаление серы из тяжелых нефтяных фракций происходит с большим трудом, чем из легких. Взаимодействие индивидуальных серосодержащих соединений различных классов с водородом в условиях гидроочистки происходит по реакции первого порядка. При гидроочистке нефтяной фракции входящие в ее состав индивидуальные соединения также реагируют по первому порядку, однако по мере удаления наиболее реакционноспособных соединений константа скорости реакции уменьшается, и в ряде случаев экспериментальные данные по изменению содержания серы в процессе гидроочистки фракции лучше описываются уравнением второго порядка. Порядок реакции по водороду может быть различным в зависимости от условий гидроочистки и свойств сырья. Кажущаяся энергия активации гидрирования серосодержащих соединений на обычных катализаторах гидроочистки в интервале 350—425°С составляет 46—48 кДж/моль. По-видимому, во всех случаях в этом температурном интервале реакция протекает во внутридиффузион-ной области.
Превращения азоторганических соединений. Азот в нефтепродуктах находится в основном в гетероциклах — в виде производных пиррола и пиридина.
Гидрогенолиз связи С—N протекает труднее, чем связи С—S, поэтому в процессах гидроочистки азот удалить сложнее, чем серу. Легче всего гидрируются амины:
Анилин, содержащий аминогруппу, связанную с ароматическим кольцом, гидрируется значительно труднее:
н2
C6H5NH2 —СбНв-|-ЫНз.
Хуже всего удаляется азот из циклических структур. Пиррол гидрируется до бутана и аммиака:
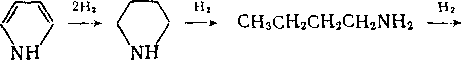
—> CH3CH2CH2CH3 + NH3.
Пиридин превращается в пентан и аммиак по схеме:
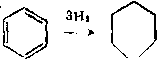
Н >
N
J -- >¦ CH3CH2CH2CH2CH2NH2 - > NH
- > CHaCHsCHsCHjCHs + NHa.
Так как сопряженная электронная система и молекуле пири-
дина значительно более устойчива, чем в молекуле пиррола,, пиридин гидрируется труднее, чем пиррол.
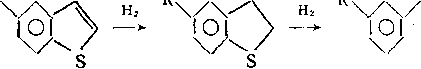
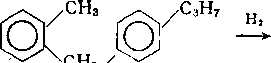
Гидрирование бициклических и полициклических аренов начинается с кольца, содержащего гетероатом:
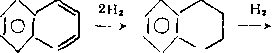
N NH
/4/CH2CH2CH2NH2 Нг /Ч/С3Н7
—* 101 —^ 101 +NH3-
В присутствии обычных катализаторов гидроочистки достигается практически полное гидрирование азотсодержащих соединений.
Превращения кислородсодержащих и металлорганических соединений. Кислород в среднедистиллятных фракциях нефтепродуктов может быть представлен соединениями типа спиртов, эфиров, фенолов и нафтеновых кислот. В высококипящих фракциях кислород находится в основном в мостиковых связях и в циклах молекул. Наибольшее количество кислородсодержащих соединений концентрируется в смолах и асфальтенах.
При гидрогенизации кислородсодержащих соединений образуются соответствующие углеводороды и вода:
R—СООН ^ R—СНз + 2Н20;
rc6h4oh RC6H5 + H20.
Смолы и асфальтены превращаются в более низкомолекулярные соединения.
Гидроочистка от кислородсодержащих соединений протекает в тех же условиях, что и удаление сернистых примесей. В присутствии обычных катализаторов гидроочистки кислородсодержащие соединения удаляются практически нацело.
Металлорганические соединения, присутствующие в нефтяных фракциях, разлагаются на активных катализаторах с выделением свободного металла, являющегося каталитическим ядом: Гидроочистка позволяет удалять большую часть металлорганических соединений (75—95 %)•
Превращения углеводородов. В условиях процесса гидроочистки алканы и циклоалканы не реагируют. Алкены, алкадие-ны и частично полициклические арены подвергаются гидрированию. В присутствии катализаторов гидроочистки алкадиены гидрируются до алканов при температуре 300—350 С под давлением водорода 0,5—2 МПа. Алкены гидрируются в более
жестких условиях —температура 350—400 °С, давление 2— 3 МПа:
н2
RCH = СН2 —> RCH2CH3.
Полициклические арены гидрируются при той же температуре, что и алкены, но под более высоким давлением — до 3— 7 МПа:
Если сопоставить энергии связей, то должны легче расщепляться и гидрироваться связи л—С—С, затем С—S, слабые <у—С—С, затем С—N и С—О. Усредненные данные по энергиям этих связей представлены ниже:
| Связь |
Усредненная энергия, кДж/моль |
| я—С—С (алкен) |
167 |
| а—С—С (слабая) | 294 |
|
С—S | 272 |
|
С—N | 335 |
|
С-0 | 377 |
Однако в присутствии катализаторов при одинаковом строении соединений устойчивость связей относительно гидрирования возрастает в ряду:
С — S<C — 0<С — N < ст — С — С.
Это явление обусловлено тем, что разрыв связей С—S, С—N и С—О облегчается вследствие их более прочной хемосорбции на катализаторе за счет электронодонорной способности атомов S, N и О.
Катализаторы процесса. В процессе гидроочистки используют катализаторы, стойкие к отравленщр различными ядами. Это оксиды и сульфиды металлов переменной валентности: Ni, Со, Mo, W на оксиде алюминия с другими добавками. В большинстве современных процессов гидроочистки используют алю-мокобальтмолибденовые (АКМ) или алюмоникельмолибденовые (АНМ) катализаторы. Эти катализаторы содержат 10—14 % МоОз и 2—3 % промотора (СоО или NiO) на активном 7-AI2O3. На стадии пусковых операций или в начале сырьевого цикла катализаторы гидроочистки подвергают сульфидированию (осернению) в токе H2S и Нг; активность катализаторов при этом увеличивается.
Кроме сероводорода используют также другие серосодержащие соединения, легко гидрирующиеся до H2S, которые дозируют в сырьевой поток. Содержание серы, связанной с катализатором, составляет 4—6 %.
Промышленные катализаторы обладают высокой избирательностью. В присутствии АКМ-катализатора с высокой скоростью протекают реакции разрыва С—S-связей, он достаточно-активен в реакциях насыщения алкенов, разрыва связей С—N и С—О. Расщепления связей С—С не происходит. Этот катализатор практически пригоден для гидроочис'гки любых нефтяных фракций. АНМ-катализатор значительно более активен в реакциях насыщения полициклических аренов и гидрирования азотистых соединений, поэтому его рекомендуют для очистки тяжелого высокоароматизированного сырья каталитического крекинга. Алюмоникель- или алюмокобальтвольфрамовые катализаторы (АНВ или АКВ) предназначены для глубокого гидрирования азотсодержащих и ароматических соединений в процессах гидрогенизационной очистки парафинов, гидрирования масел и др. Пока эти катализаторы используют мало, но они найдут большое применение при переработке тяжелого сырья. В последние годы широкое распространение получают катализаторы на цеолитной основе, отличающиеся повышенной активностью и стабильностью. В процессе работы катализатор закок-совывается и теряет активность. Для ее восстановления катализатор подвергают регенерации, выжигая кокс с поверхности при температуре до 530 °С.
Вопрос о природе активности катализаторов гидроочистки пока далек от окончательного решения. После сульфидирова-ния АКМ-катализатор содержит как оксиды, так и сульфиды молибдена. Суммарная стехиометрия соответствует образованию фазы MoOxSу {х + у = 3), а не Мо02 + MoS2, причем молибден присутствует в различных валентных состояниях (от 4+ до 6+), прочно связанных с носителем. Оксиды молибдена и вольфрама являются «-полупроводниками (электронные). Их активность может быть обусловлена наличием на поверхности свободных электронов, способствующих адсорбции, гемолитическому распаду и гидрированию адсорбированных органических молекул.
В оксидных АКМ-катализаторах наблюдаются внедрение ионов Со2+ в А1203 и сильное электронное взаимодействие молибдена с А120з, что, по-видимому, способствует увеличению числа активных центров гидрирования — дегидрирования.
Сульфидные катализаторы являются ^-полупроводниками (дырочные). Дырочная проводимость этих веществ обусловлена примесью серы. Поэтому формула сульфида вольфрама WS2, 2, а не WS2. Под влиянием дырок на поверхности катализаторов возможно протекание гетеролитических процессов в органи-
R
Н

+ R
В сульфидированных контактах кобальт обнаруживается в виде фазы C0M0S2.
Таким образом, сульфидированные катализаторы гидроочи--стки являются бифункциональными и могут ускорять как ионные, так и радикальные процессы.
Макрокинетика процесса. Скорость протекания реакций гидроочистки нефтяных фракций зависит от химической природы и физических свойств сырья, типа катализатора, парциального давления водорода, объемной скорости подачи сырья, температуры и других факторов.
С повышением температуры скорость реакций гидрирования возрастает. Однако верхний предел температуры ограничен (400—420 °С), что связано с неблагоприятным термодинамическим равновесием гидрирования тиофенов и, вероятно, также хинолина и бензохинолина. Кроме того, повышение температуры способствует реакциям гидрокрекинга, дегидрирования поли-циклических циклоалканов и коксообразованию. В зависимости от качества исходного сырья и требуемого качества очищенного продукта гидроочистку проводят при температуре 250—420 °С.
Скорость газофазной реакции (при гидроочистке легких нефтяных фракций) возрастает с увеличением парциального давления водорода примерно до 2—3 МПа и далее почти не меняется. В жидкофазном процессе (при очистке высококипящих фракций) повышение давления водорода до очень высоких значений увеличивает скорость реакции, ускоряя транспортирование водорода через пленку жидкости к поверхности катализатора. Предел повышения давления обычно ограничивается удорожанием оборудования и составляет 7—8 МПа.
Объемная скорость подачи сырья зависит от содержания и типа гетероатомных соединений в сырье, от технологии получения сырья (первичное, вторичное) и требуемой глубины очистки. Обычно она колеблется в очень широких пределах — •от 0,5 до 10 ч-1. Гидроочистку сырья с высоким содержанием тиофенов проводят с меньшей объемной скоростью, чем сырья, содержащего серу в виде меркаптанов и сульфидов. Низкая объемная скорость требуется также для переработки тяжелого сырья и сырья вторичного происхождения из-за высокого содержания непредельных и полициклических ареной, а также трудностей удаления высокомолекулярных гетероатомных соединений.
Гидроочистка в промышленности. В промышленности гидро->чистку нефтяных фракций проводят при 380—420 °С под даванием 2,5—4 МПа в присутствии АКМ (или АНМ) катализаторов. Соотношение водород : сырье в м3 обычно составляет (300—600) : 1. В этих условиях происходят полное удаление ге-героатомов, металлов и гидрирование алкенов; в тяжелых фракциях частично гидрируются полициклические арены. Гидроочи-:тке подвергают любые фракции, а также нефтяные остатки..
Гидроочистка бензиновых фракций. Гидроочи^-стку бензинов проводят в основном с целью подготовки сырья для процесса риформинга. Так как катализатор риформинга: отравляется гетероатомными соединениями, то глубина очистки, должна быть очень высокой: остаточное содержание серы
в сырье риформинга на платиновом катализаторе не может быть выше 4—5 млн-1 (мг/кг), на биметаллических катализаторах —
1 млн-1 (мг/кг). Очистка от гетероатомных и металлоргани-ческих соединений бензинов прямой перегонки нефти происходит обычно при температуре 320—360 0С под давлением 3—
5 МПа, циркуляции водородсодержащего газа 200—500 м3/м3 сырья и объемной скорости 5—10 ч-1. При очистке бензинов вторичного происхождения (каталитического крекинга, термических процессов) кроме удаления гетероатомов ставится задача селективного гидрирования алкенов при сохранении аренов. Для ее осуществления процесс проводят с меньшей объемной скоростью (0,5—5 ч-1) и при большем отношении водорода к сырью (400—600 м3 Нг/м3 сырья).
Гидроочистка керосиновых фракций. Целью* процесса является получение малосернистого реактивного топлива, осветительного керосина или растворителя. Процесс проводят практически в тех же условиях, что и гидроочистку прямогонного бензина. В товарном реактивном топливе содержание серы допускается не более 0,1 %, а в осветительных керосинах— 0,05—0,1 %.
Другой важной характеристикой реактивных топлив является содержание аренов, которое не должно превышать 10— 16% Для топлива Т-6 и 18—22% для топлив Т-1, Т-2, Т-8 и РТ. В керосинах прямой перегонки содержание аренов составляет 14—30%, а в легком газойле каталитического крекинга — 60—70 %. Особенно нежелательны примеси би- и полициклических аренов. Если ставится задача понизить концентрацию-аренов, то процесс проводят на более активном катализаторе под давлением до 7 МПа.
Гидроочистка дизельных топлив. Повышенный интерес к развитию процессов гидроочистки средних дистиллятов в последние годы связан с увеличением объема переработки сернистых и иысокосернистых нефтей и широкой дизелизацией транспортных средств. В СНГ в настоящее время гидроочистке подвергают более 80% дизельных фракций. При этом выпуск дизельных топлив с содержанием серы 0,2—0,5 % составляет примерно 90 %. Прямогонные дизельные фракции подвергают гидроочистке без заметного изменения их группового и фракционного состава на АКМ-катализаторах при температуре 350— 400 °С под давлением 3—4 МПа, объемной скорости подачи сырья 2—5 ч-1 и циркуляции водородсодержащего газа 300— 600 м3/м3 сырья. Степень гидрообессеривания составляет 85—95 %.
В связи с увеличением потребности народного хозяйства в дизельном топливе приобретает особую актуальность получение высококачественных дизельных топлив из дистиллятов вторичного происхождения: продуктов каталитического крекинга, замедленного коксования, висбрекинга. Это сырье отличается от прямогонного повышенным содержанием серы, азота, смол, алкенов и аренов. Для его очистки процесс проводят при более низкой объемной скорости — около 1 ч-1, под более высоким давлением водорода-—примерно 5 МПа. Дизельные топлива вторичного происхождения характеризуются низкими цетано-выми числами, обусловленными высокой концентрацией аренов.
Для повышения цетановых характеристик необходимо гидрирование большей части аренов, осуществляемое на активных катализаторах при температуре около 400°С под давлением водорода до 10 МПа.
Гидроочистка вакуумных дистиллятов. Вакуумные дистилляты (вакуумные газойли) являются сырьем процессов каталитического крекинга, гидрокрекинга, получения электродного кокса. Для повышения выхода и улучшения качества продуктов этих процессов и уменьшения загрязнения окружающей среды оксидами серы все большая доля вырабатываемых вакуумных газойлей подвергается гидроочистке.
Гидроочистка вакуумного газойля первичной перегонки нефти не представляет значительных трудностей. Проводят ее в условиях и на оборудовании, аналогичных для гидроочистки средних дистиллятов: температура 360—410°С, давление 4—
5 МПа, объемная скорость подачи сырья 1—1,5 ч-1. При этом достигается 90—94 % степень гидрообессеривания; содержание азота снижается на 20—25 %; металлов — на 75—85 %; аренов— на 10—12%; коксуемость — на 65—70 %. Тяжелые вакуумные газойли вторичного происхождения (замедлеииого коксования, висбрекинга) характеризуются высоким содержанием серы, азота, алкенов, аренов, смол. Такие газойли рекомендуют перерабатывать в смеси с первичными, добавляя их в количестве до 30 %.
Нели тяжелые газойли вторичных процессов предназначены в качестве сырья для получения технического углерода, то в процессе их гидроподготовки необходимо удалить только соединения серы и азота, не затрагивая аренов. Такую задачу решают подбором условий и катализаторов.
Гидроочистка масел и парафинов. Гидроочистка масляных фракций служит для улучшения таких свойств, как стабильность, цвет, коксуемость, путем удаления гетероатомных полициклических и смолистых веществ. Процесс более технологичен по сравнению с сернокислотной и контактной доочисткой. Гидроочистку масляных фракций проводят при температуре 300—325 °С, 4 МПа на АКМ- и АНМ-катализаторах. Перспективным является алюможелезомолибденовый катализатор с промоторами, на котором гидроочистка масел успешно протекает при температуре 225—250 °С под давлением 2,7— 3,0 МПа.
Гидроочистка парафинов, церезинов и петролатумов также снижает содержание в них сероорганических соединений, алкенов, смол, улучшает цвет и стабильность. Процесс проводят в условиях, близких к гидроочистке масел. Кроме АКМ- и АНМ-катализаторов используют также алюмохроммолибдено-вые и никельвольфрамжелезные сульфидированные катализаторы.
Гидроочистка нефтяных остатков. Выход нефтяных остатков (мазутов, гудронов) достигает 45—55 % на нефть. Одним из путей углубления переработки нефти и увеличения отбора светлых нефтепродуктов является каталитическая переработка нефтяных остатков. По сравнению с дистиллятным сырьем остатки характеризуются более высоким содержанием серо-, азот- и металлорганических соединений, смол, асфальтенов, золы. Для подготовки нефтяных остатков к каталитической переработке предложен ряд методов непрямого гидрообессери-вания, которые заключаются в вакуумной перегонке мазута и деасфальтизации выделившегося гудрона с последующей гидроочисткой вакуумного газойля и деасфальтизата. Если очищенные продукты не смешивать с остатком деасфальтизации, то содержание серы в котельном топливе снижается почти на порядок (до 0,2—0,3%). При смешении очищенных продуктов с остатком содержание серы в топливе составляет 0,4—1,4 %.
В современных схемах нефтеперерабатывающих заводов чаще применяют прямое гидрообессеривание мазута или раздельную каталитическую переработку вакуумного дистиллята и гудрона. Прямое гидрообессеривание мазута проводят при следующих условиях: температура 370—427°С, давление 10— 15 МПа, объемная скорость подачи сырья 0,5 ч 1 па АКМ-ката-лизаторе. Выход мазута с содержанием серы до 0,3 % составляет 97—99 %. Одновременно с обессеривакием происходят удаление азота, смол, асфальтенов и частичная деструкция сырья.
Гидроочистка гудронов представляет собой более сложную задачу, чем гидроочистка мазутов. Эффективная переработка такого сырья возможна только при его предварительной деметаллизации или деасфальтизадии. Основным недостатком всех процессов прямого гидрообессеривания остатков является быстрая дезактивация катализатора из-за отложений кокса и металлов. При отравлении коксом активность катализатора восстанавливается путем регенерации. При отравлении металлами (V, Ni) окислительная регенерация не восстанавливает актив-лости катализатора. Введение в технологию гидроочистки остатков стадии деметаллизации позволяет снизить расход катализаторов в 3—5 раз.
14.3. ГИДРОКРЕКИНГ
Гидрокрекинг — каталитический процесс, предназначенный для получения светлых нефтепродуктов (бензина, керосина, дизельного топлива),.а также сжиженных газов С3 — С4 при переработке под давлением водорода нефтяного сырья, имеющего молекулярную массу более высокую, чем получаемые целевые продукты.
Гидрокрекинг позволяет получать широкий ассортимент нефтепродуктов практически из любого нефтяного сырья путем подбора соответствующих катализаторов и условий и является одним из наиболее эффективных и гибких процессов нефтепереработки.
Химические основы процесса. Характеристики продуктов гидрокрекинга в очень сильной степени определяются свойствами катализатора — его гидрирующей и кислотной активностью. Катализаторы гидрокрекинга можно разделить на имеющие высокую гидрирующую и относительно низкую кислотную активность и имещие относительно невысокую гидрирующую и высокую кислотную активность.
Превращения алканов. На монофункциональных гидрирующих катализаторах, не обладающих кислотными свойствами, протекает гидрогенолиз алканов путем диссоциации одной из С—С-связей на катализаторе и последующего насыщения осколков водородом по схеме:
г
^п^2п + 2 ^2 v^т^2т + 2 ^п—т^2{п—т)+2-
Скорость разрыва различных С—С-связей главным образом зависит от выбранных катализаторов: на платине скорости гидрогенолиза всех С—С-связей близки, на никеле быстрее расщепляются концевые С—С-связи с образованием метана. Высокую активность в процессе гидрогенолиза С—С-связей проявляют также некоторые цеолиты. Реакция протекает гомолити-чески, с участием электронов катализатора.
На кислотных н бифункциональных катализаторах алканы подвергаются крекингу и изомеризации по гетеролитическому механизму. Вначале на активных центрах гидрирования — дегидрирования происходит дегидрирование углеводородов с образованием алкенов. Алкены далее легко превращаются в карб-катионы на кислотных центрах катализатора и инициируют цепной карбкатионный процесс, аналогичный каталитическому крекингу. Как и при каталитическом крекинге, скорость гидрокрекинга возрастает с увеличением молекулярной массы алканов. Изоалканы с третичными углеродными атомами подвергаются гидрокрекингу, как и каталитическому крекингу, со значительно большей скоростью, чем неразветвленные алканы.
Основные отличия каталитического крекинга от гидрокрекинга заключаются в том, что общая конверсия алканов при гидрокрекинге выше, чем при каталитическом крекинге. Это обусловлено легкостью образования алкенов на гидрирующих — дегидрирующих центрах катализаторов гидрокрекинга. В результате наиболее медленная и энергоемкая стадия цепного процесса — инициирование цепи, — определяющая скорость всего процесса, при гидрокрекинге протекает быстрее, чем при каталитическом крекинге. Продукты гидрокрекинга имеют предельный характер. Катализаторы гидрокрекинга практически не закоксовываются, так как алкены подвергаются быстрому гидрированию и не успевают вступать в дальнейшие превращения по пути полимеризации и уплотнения.
Соотношение выхода продуктов гидрокрекинга алканов определяется соотношением скоростей изомеризации карбкатионов, их распада и стабилизации. На катализаторах с высокой кислотной и умеренной гидрирующей активностью гидрокрекинг идет с высокой скоростью, причем образуется много низкомолекулярных изоалканов. Это объясняется высокой скоростью изомеризации и распада карбкатионов на сильных кислотных центрах. На катализаторах с высокой гидрирующей и умеренной кислотной активностью степень превращения алканов невелика, что, вероятно, обусловлено быстрым насыщением карбкатионов при реакциях типа
1ГА-+ГН-Н —> RH + HA + Г,
где А- — анион катализатора НА; Г — каталитический центр гидрирования — дегидрирования.
В результате подобных превращений главными продуктами становятся алканы с большим числом атомов углерода и меиее изомсризованные, чем на катализаторах с высокой кислотностью.
Превращения циклоалканов. Превращения незамещенных и метилзамсщенных моноциклоалканов в присутствии гидрирующих катализаторов заключаются главным образом в гидрогено-лизе — расщеплении кольца по связям, определяемым катализатором, за которым следует насыщение обоих концов образовавшегося осколка. На бифункциональных катализаторах, с относительно низкой кислотной активностью разрыв происходит по С—С-связи преимущественно в р-положении по отношению к заместителю, что объясняется образованием третичного карбкатиона и его распадом по (3-связи. Например, при температуре 250—270 °С под давлением водорода 2,1 МПа на алюмо-платиновом катализаторе метилциклопентан превращается в 2-метилпентан, 3-метилпентан и я-гексан в соотношении 7:2:1 по следующей схеме:
СН3СН2СН2СН (СНз) СН;
(3-связь
Нг Y-связь
о-
СН3СН2СН (СНз)СН2СНз
>—СН3
а-связь
¦СН3СН2СН2СН2СН2СН3
На катализаторах с высокой кислотностью и низкой гидрирующей активностью протекают в основном реакции изомеризации шестичленных циклоалканов в пятичленные и по положению заместителей. Расщепление кольца происходит лишь в небольшой степени. Высокая устойчивость циклоалкановых колец при гидрокрекинге на катализаторах с высокой кислотной активностью объясняется тем, что для образующегося при распаде кольца карбкатиона обратная реакция протекает быстрее, чем дальнейший распад по (3-правилу или стабилизация за счет отрыва гидрид-иона от исходной молекулы
СНз
СНзСН = СН2 + СН2С = СН2
g-распад
СНз
СНз
СНз
СНз
СНз
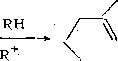
СНз
Циклоалканы с длинными алкильными боковыми цепями в условиях гидрокрекинга подвергаются главным образом изомеризации и распаду алкильных заместителей. Бициклические циклоалканы превращаются в моноциклические с высоким выходом производных пентапа.
Превращения алкенов. Алкены на кислотных центрах катализатора превращаются в карбкатиоиы и вступают в реакции, характерные для этих частиц. Они изомсризуются и подвергаются распаду по p-нранилу (см. гл. 13). Одновременно на гидрирующих центрах происходит насыщение алкенов — как исход-
R_CH = CH2K-ii RCHCH3 К:изомеРиз^ия1Р-распад>
I Г;Н2
1—> rch2ch3
—> R+ + Изоалкен меньшей молекулярной массы (или алкен)
1 к; -н+ v
Сумма низкомолекулярных алкенов и изоалкенов ]г;Н2
Сумма низкомолекулярных алканов и изоалканов
где К — кислотный центр катализатора; Г — активный центр гидрирования — дегидрирования.
Соотношение скоростей реакций гидрирования алкенов и превращения их по ионному пути определяет активность катализатора. На катализаторах с высокой кислотной активностью скорость изомеризации и распада ионов выше скорости насыщения. Это приводит к образованию низкомолекулярных разветвленных соединений, главным образом изобутана. На катализаторах с высокой гидрирующей активностью происходит интенсивное насыщение алкенов, в результате чего образуются
алканы с большой молекулярной массой и незначительной сте
пенью изомеризованности. Полнота гидрирования алкенов зависит также от их молекулярной массы и режима процесса. Скорость гидрирования уменьшается с увеличением числа атомов углерода в молекуле непредельного соединения. Октилен гидрируется почти в два раза медленнее, чем этилен.
Превращения аренов. На катализаторах с высокой гидрирующей и низкой кислотной активностью происходит насыщение ареновых колец. Арены гидрируются труднее алкенов. Присоединение водорода к любой двойной связи протекает с выделением теплоты, гидрирование же бензола в 1,2-дигидробензол эндотермично. Дальнейшее гидрирование 1,2-дигидробензола идет легко и экзотермически (цифры — энергия связи в кДж/моль):
н, ib н2
CeHj — >- СвН8 — * СвНю —> СвН,2.
-21 | 110 -| 119
Гомологи бензола гидрируются труднее, чем бензол, вследствие пространственных трудностей при адсорбции иа поверхности катализатора. Если скорость гидрирования бензола принять за единицу, то для гомологов скорости будут примерно
Изопропилбеизол 0,3
1,3,5-Трнметилбензол 0,2
Толуол
Этнлбеизол
Гидрирование первого кольца в полициклических аренах происходит быстрее, чем гидрирование бензола, что объясняется неравномерным распределением я-электронной плотности в по* лициклических соединениях. Например, скорость превращения антрацена в 9,10-дигидроантрацен в 3,3 раза выше, чем скорость гидрирования бензола. Основной принцип при гидрировании конденсированных ароматических систем — последовательность насыщения бензольных колец водородом, причем по мере насыщения скорость реакции снижается. Например, если относительную скорость гидрирования антрацена в 9,10-дигидроантрацен с насыщением у-углеродных атомов в среднем кольце принять равной единице, то присоединение следующего моля водорода идет со скоростью 0,94, а скорость гидрирования последнего кольца составляет 0,01 (цифры — относительная скорость реакции) :
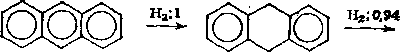
Наряду с последовательным гидрированием ароматических колец возможно расщепление образовавшихся насыщенных колец и выделение алкилзамещенных аренов:
сн2/
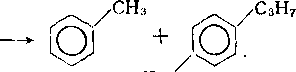
НзС
Алкилбензолы на катализаторах с высокой гидрирующей активностью подвергаются дальнейшему гидрогенолизу, в основном с последовательным отщеплением метана:
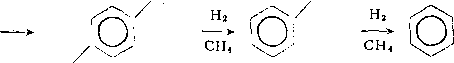
НзС
На катализаторах с высокой кислотной и низкой гидрирующей активностью превращения аренов во многом аналогичны каталитическому крекингу (см. гл. 13). Незамещенные моноциклические арены стабильны. Метил- и этилбензолы вступают в реакции изомеризации по положению заместителей и диспро-порционирования. Алкилбензолы с более длинными цепями де-алкилируются. Образовавшиеся алкильные карбкатионы после изомеризации подвергаются p-распаду и насыщаются по схеме, описанной для гидрокрекинга алканов, с образованием смеси низкомолекулярных алканов нормального и изостроения. Алкилбензолы, кроме того, могут превращаться в тетралин и индан по схеме:
CH2CH2CH2CH2R
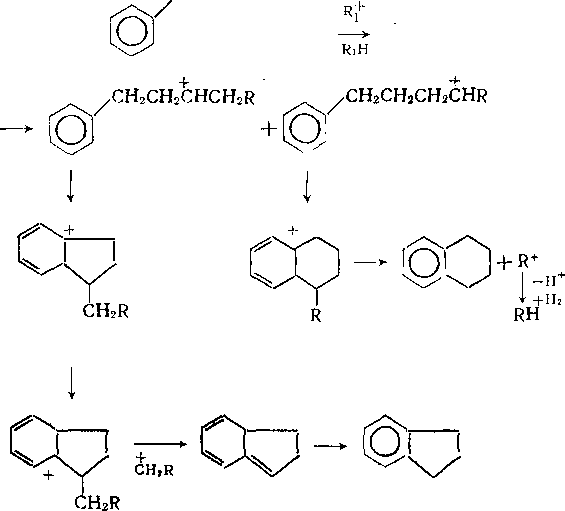
Полициклические ареиы на катализаторах с высокой кислотностью гидрируются до моноциклических с различными алкильными заместителями и далее расщепляются, как алкилбензолы. В результате гидрокрекинга полициклических аренов в значительном количестве образуются также производные тетралина и индана.
Сравнение скоростей гидрокрекинга углеводородов различных классов свидетельствует о том, что гидрирование полицик-лических структур до углеводородов, содержащих по одному ароматическому или алициклическому кольцу, происходит быстро. Гидрирование аренов и циклоалканов с разрушением последнего кольца протекает сравнительно медленно. Относительно медленно проходит также гидрокрекинг алканов. Таким образом, в продуктах реакции накапливаются производные моцоциклических аренов и циклоалканов, а также алканы, преимущественно разветвленные.
Катализаторы процесса. Ассортимент катализаторов гидрокрекинга достаточно широк, что объясняется разнообразием назначений процесса. Обычно они состоят из следующих компонентов: кислотного, гидрирующе-дегидрирующего и связующего, обеспечивающего механическую прочность и пористую структуру.
В качестве кислотного компонента, выполняющего крекирующую и изомеризующую функции, используют цеолиты, оксид алюминия, алюмосиликаты. Для усиления кислотности в катализатор вводят галоген, дополнительные оксидные добавки или проводят предварительное деалюминирование или дека-тионирование цеолита.
Гидрирующим компонентом обычно служат металлы VIII группы (Pt, Pd, Ni, Со, Fe), а также оксиды или сульфиды некоторых металлов VI группы (Mo, W). Для повышения активности перед использованием металлы VIII группы восстанавливают водородом, а оксидные молибден- и вольфрамсодержащие катализаторы сульфидируют; кроме того, для активирования катализаторов используют также разнообразные промоторы. В качестве промоторов наиболее известны рений, родий, иридий, редкоземельные элементы — для металлов VIII группы— и оксиды кобальта и никеля — для катализаторов на основе металлов VI группы. Функции связующего часто выполняют кислотный компонент (оксид алюминия, алюмосиликаты), а также оксиды кремния, титана, циркония, магний- и цирко-нийсиликаты.
Сульфиды и оксиды молибдена и вольфрама с промоторами являются бифункциональными катализаторами: они активны как в реакциях гидрирования — дегидрирования, так и в кислотнокаталитических реакциях (см. катализаторы гидроочистки).
Оптимальные результаты гидрокрекинга достигаются при использовании катализаторов с высокой кислотной и умеренной гидрирующей активностью.
Большинство катализаторов, содержащих металлы VIII группы, легко отравляются каталитическими ядами, к которым относят элементы V группы (N, Р, As, Sb, Bi) и часть элементов VI группы (О, S, Se, Те). Поэтому гидрокрекинг сырья, содержащего значительное количество гетеро- и металлорганических соединений, обычно проводят в две ступени. На первой ступени в основном проходят гидроочистка и неглубокий гидрокрекинг полициклических аренов. Катализаторы этой ступени идентичны катализаторам гидроочистки. Они содержат оксиды и сульфиды никеля, кобальта, молибдена и вольфрама на активном оксиде алюминия, алюмосиликате или цеолите. На второй ступени подготовленное сырье, содержащее не более 10-2 % серы и не более 10~4 % азота, перерабатывается на катализаторах, включающих палладий или платину на цеолите Y.
В одноступенчатом гидрокрекинге дистиллятных фракций используют бифункциональный катализатор, в котором гидрирующую функцию выполняют элементы платиновой группы (0,1—3,0 %), а также никель (2—10 %) или композиции никеля (кобальта) в количестве 2,5—5 % и молибдена (вольфрама) 5—15% в сульфидной форме. В качестве кислотного компонента выступают цеолит, оксид алюминия или алюмосиликат.
В процессе селективного гидрокрекинга в качестве катализаторов применяют модифицированные цеолиты (модернит, эрионит и др.) со специфическим молекулярно-ситовым действием: поры цеолитов доступны только для молекул алканов нормального строения. Гидрирующе-дегидрирующие функции в таких катализаторах выполняют те же металлы и соединения, что и в одноступенчатом гидрокрекинге.
Макрокинетика процесса. Превращение сырья в условиях процесса гидрокрекинга идет по следующим направлениям. В первую очередь гидрогенолизу подвергаются неуглеводородные соединения, вследствие чего из сырья удаляются гетероатомы в виде Н20, NH3 и H2S. Одновременно происходит гидрирование углеводородов, имеющих ненасыщенный характер. Полициклические арены и циклоалканы гидрируются в замещенные моноциклические. Алканы подвергаются изомеризации и расщеплению. Значительно труднее (в более жестких условиях или в присутствии более активных катализаторов) происходит насыщение последнего ароматического кольца и гидро-генолиз алканов и моноциклоалканов. Соотношение скоростей различных реакций гидрокрекинга легкого газойля каталитического крекинга на катализаторе с высокой кислотной активностью щэи 10,5 МПа приведено на рис. 14.1.
Расщепление и изомеризация являются типичными реакциями первого порядка. Гидрирование и деструктивное гидрирование— реакции второго порядка. Однако в связи с большим избытком водорода в системе их также описывают уравнениями первого порядка. Таким образом, гидрокрекинг в целом можно представить кинетическими уравнениями реакций
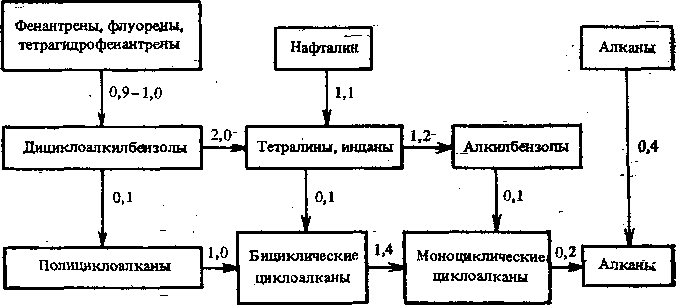
Рис. 14.1. Схема превращения углеводородов в условиях процесса гидрокрекинга
первого порядка с торможением реакции образующимися продуктами. Кажущаяся энергия активации гидрокрекинга вакуумного газойля в температурном интервале 380—420 °С составляет 140—250 кДж/моль.
Тепловой эффект гидрокрекинга определяется соотношением реакций гидрирования и расщепления. Обычно отрицательный тепловой эффект расщепления перекрывается положительным тепловым эффектом реакции гидрирования. В общем тепловой эффект процесса гидрокрекинга может изменяться от —208 до 834 кДж/кг сырья. Расход водорода на реакцию зависит от назначения процесса, используемого сырья, катализатора, режима процесса и других факторов. Водородсодержащий газ подается в количестве 500—2000 м3/м3 сырья. Чем легче продукты, получаемые из данного сырья, тем больше расход водорода и тем выше должно быть соотношение водород: сырье.
Оптимальная температура проведения гидрокрекинга обычно 300—425°С. При более низкой температуре реакции протекают с малой скоростью. Чрезмерное повышение температуры ограничивается термодинамическими факторами реакции гидрирования и увеличением скорости коксообразования. Кроме того, при повышенной температуре значительно ускоряются реакции распада, идущие с наибольшей энергией активации, в результате чего увеличивается выход легких фракций и газа. Вследствие желательности проведения процесса при минимальной температуре объемная скорость подачи сырья при гидрокрекинге низка: 0,5—2,0 ч-1. Давление, минимально необходимое для переработки легких газойлей при 400—425°С, составляет примерно 7 МПа. При давлении менее 5 МПа начинается интенсивное лакоксопыпапие катализатора. Для тяжелых газойлей и тем более остаточного сырья с целью предотвращения обратной реакции дегидрирования циклоалкановых колец в полицик-лических системах требуется более высокое давление (до 20— 30 МПа).
Гидрокрекинг в промышленности. По целевому назначению реализованные в промышленности процессы гидрокрекинга можно разделить на следующие:
1) гидрокрекинг бензиновых фракций с целью , получения сжиженного нефтяного газа, углеводородов С4—С5 изостроения для нефтехимического синтеза и легкого высокооктанового компонента автомобильных бензинов;
2) гидрокрекинг средних дистиллятов (прямогонных И вторичного происхождения) с температурой кипения 200—350 °С с целью получения бензинов и реактивных топлив;
3) гидрокрекинг атмосферного и вакуумного газойлей, газойлей коксования и каталитического крекинга с целью получения бензинов, реактивного и дизельного топлив;
4) гидрокрекинг тяжелых нефтяных дистиллятов с целью получения реактивных и дизельных топлив, смазочных масел, малосернистых котельных топлив и сырья для каталитического крекинга; ¦
5) селективный гидрокрекинг бензинов с целью повышения октановых чисел; реактивных и дизельных топлив с целью снижения температуры застывания; масляных фракций — для улучшения цвета, стабильности и снижения температуры застывания;
6) гидродеароматизация.
Гидрокрекинг бензиновых фракций. Разработан и нашел промышленное применение комбинированный процесс каталитического риформинга и гидрокрекинга бензинов, который в нашей стране получил название изоформинга. В этом процессе сырье — тяжелые бензиновые фракции — перед рифор-мингом подвергают гидрокрекингу, совмещенному с гидроочисткой. Продукт гидрокрекинга, очищенный от гетерооргани-ческих соединений, содержит до 20 % низкомолекулярных алканов (изокомпонента), которые отделяют ректификацией. Остаток после ректификации по сравнению с исходным сырьем имеет облегченный фракционный состав и характеризуется повышенным содержанием аренов и циклоалканов, то есть является лучшим сырьем для каталитического риформинга. Оптимальные результаты гидрокрекинга бензинов получены на ни-кель-алюмосиликатном, никель-цсолитном и никсль-молйб-ден-цеолитном катализаторах при температуре 300—350 °С, давлении 2—9 МПа, объемной скорости подачи сырья 1—2 ч~' и циркуляции водородсодержащего газа 1000—1500 м3/м3 сырья.
Октановое число изокомионента составляет 86 пунктов по исследовательскому методу. Смешением изокомпонента с ри-форматом в соотношении 3: 7 получают бензин АИ-93.
Недостатком изоформинга является большой выход газа—¦ соотношение изокомпонент: газ примерно равно 1:1. Эффективность процесса возрастает при использовании сжиженных газов для газобаллонных двигателей или при комбинировании с процессами, потребляющими изобутан (производство изопрена, метил-грег-бутилового эфира, полиизобутилена).
Другим вариантом комбинированной переработки бензиновых фракций является сочетание риформинга с гидроизомеризацией (ригиз). Сущность процесса заключается в частичном гидрировании аренов, содержащихся в бензине риформинга (риформате), в циклоалканы по схеме: арены-»-шестичленные циклоалканы-»-пятичленные циклоалканы. Гидроизомеризацию риформатов проводят на алюмоплатиновом катализаторе при температуре 250—450 °С под давлением 1,5—7 МПа. Осуществление этого процесса позволяет уменьшить количество изокомпонентов, добавляемых в неэтилированный бензин, при сохранении высокого октанового числа. Метилзамещенные циклопентаны имеют более высокие октановые числа смешения, чем бензол. Дополнительным преимуществом получаемого бензина является снижение концентрации бензола — наиболее токсичного компонента бензина (скорость гидрирования менее токсичных гомологов бензола ниже, чем бензола). Процесс связан с большим расходом водорода и экономически целесообразен на нефтеперерабатывающих заводах с избыточными ресурсами водорода.
Гидрокрекинг средних дистиллятов. Гидрокрекинг средних дистиллятов (200—350 °С) для получения бензинов и реактивных топлив изучен, но практического значения не имеет в связи с отсутствием ресурсов сырья.
Гидрокрекинг тяжелых газойлевых фракций. В промышленности реализованы варианты гидрокрекинга тяжелых газойлевых фракций, направленные на получение бензина, реактивного и дизельного топлива, а также повышение качества смазочных масел, котельного топлива и сырья каталитического крекинга и пиролиза.
Гидрокрекинг малосернистых вакуумных дистиллятов в бензин осуществляется на катализаторах, стойких к отравлению гетероатомными соединениями (сульфидные катализаторы), в одну ступень при температуре 340—450 °С под давлением водорода 10—20 МПа. Выход бензина обычно составляет 30— 40%, но может достигать 80—90 % (об.). Для переработки сырья, содержащего выше 1,5 % серы и 500—2500 млн 1 азота, применяется дмухступенчатый процесс со стадией гидроочистки па первой ступени. Вторую ступень процесса осуществляют на катализаторах, содержащих металлы VIII группы, при температуре 290—380 °С под давлением 7—10 МПа. Выход бензина достигает 70—125 % (об.) на сырье. Получающийся легкий бензин (к. к. 190 °С) используют как компонент товарного бензина. Тяжелый бензин направляют на риформинг.
Гидрокрекинг тяжелых газойлей в среднедистиллятные фракции (реактивное и дизельное топливо) также проводят по одно- и двухступенчатой схемам. Наиболее распространен одноступенчатый процесс на катализаторах, не чувствительных к ядам, при температуре 380—410 °С и давлении водорода 12—15 МПа. Режим процесса подбирают таким образом, чтобы при невысоком выходе бензина получать до 85 % реактивного или дизельного топлива. В СНГ разработан одноступенчатый процесс гидрокрекинга вакуумного газойля в Одну ступень на цеолитсодержащем катализаторе ГК-8 с получением 52 % реактивного топлива или до 70 % зимнего дизельного топлива с остаточным содержанием аренов 5—7%. Гидрокрекинг вакуумных дистиллятов сернистых нефтей проводят по двухстадийной схеме.
Включение гидрокрекинга в схемы переработки нефти обеспечивает гибкость эксплуатации предприятий. Изменяя технологический режим процесса и условия ректификации жидких продуктов, можно на одной и той же установке получать любой из перечисленных продуктов: бензин, реактивное или дизельное топливо. В табл. 14.2 в качестве примера приведены различные варианты процесса двухступенчатого гидрокрекинга тяжелого дистиллятного сырья (фракция 350—500 °С прямогонного газойля). Переход с одного варианта на другой осуществляют изменением температуры в реакторах, а также изменением режима и направления потоков в блоке разгонки продуктов гидрокрекинга.
Бензиновый вариант дает возможность получать 51 % бензина в расчете на сырье. При этом легкий бензин (фракция С5 — С6) имеет октановое число 82, а фракция С7 — Сю — 66 (по моторному методу) при содержании серы 0,01 %. Фракция С7 — Сю может быть подвергнута риформингу для повышения октанового числа. Дизельное топливо, получаемое в этом варианте (фракция 180—350 °С) с выходом 25,4 % от сырья, имеет цетановое число 50—55, содержит 0,01 % серы и застывает при температуре не выше —10 °С. Эта фракция отвечает всем требованиям стандарта на летнее дизельное топливо.
Продукты гидрокрекинга по бензиновому варианту во многом сходны с продуктами каталитического крекинга: газооб
разные продукты содержат значительное количество углеводородов Сз — С4, в жидких продуктах много разветвленных соединений. В отличие от каталитического крекинга, продукты гидрокрекинга имеют насыщенный характер и практически не содержат гетероатомных соединений. Газойли гидрокрекинга,
Таблица 14.2. Продукты гидрокрекинга вакуумного дистиллята сернистой нефти
Условия процесса: давление 15 МПа; объемная скорость в каждой ступени
1 ч-l; кратность циркуляции водородсодержащего газа 1000—1700 м3/м3 сырья; температура I ступени 420 °С, катализатор АКМ; температура П ступени 320—425 °С, катализатор Ni или Pt на алюмосиликате.
| Целевой продукт (вариант отбора) | ||||
|
Показатели | бензин |
реактив ное топливо |
дизельное топливо |
бензин и дизельное топливо |
| Расход 100 %-го водорода, % |
4,10 | 3,82 | 2,40 | 3,63 |
|
Выход в расчете на сырье, %: | ||||
| сухой газ (+потерн) |
6,50 | 7,62 | 5,70 |
6,83 |
|
сероводород | 2,30 |
2,30 | 2,30 | 2,30 |
| фракция Сз—С4 (сжижен- | 10,60 |
10,20 | 4,30 | 8,40 |
| ныи газ) бензиновые фракции | ||||
|
Сб-—Се | ¦—¦ |
26, | — |
- |
|
С;—Св (легкий бензин) |
17,62 | — |
2,60 | 9,20 |
|
С7—Сю | 33,40 |
— | 12,80 |
20,40 |
|
керосиновая фракция | ||||
|
120—240 °С | — |
41,50 | — |
— |
|
дизельные фракции | ||||
|
180—350 °С | 25,40 |
— | 66,90 | 47,00 |
| 240—350 °С |
— | 10,00 |
— | — |
| газойлевая фракция | ||||
| 350—450 °С | 8,30 |
10,00 | 7,90 | 9,50 |
кроме того, менее ароматизированы, чем газойли каталитического крекинга.
Реактивно-топливный вариант позволяет получать до 41,5 % фракции 120—240 °С, отвечающей требованиям стандарта на реактивное топливо. При двух других вариантах, имеющих дизельно-топливное направление, можно получать 47 и 67 % дизельного топлива с цетановым числом 50.
Перспективным направлением гидрокрекинга является переработка масляных фракций (вакуумных дистиллятов и де-асфальтизатов). Глубокое гидрирование масел позволяет повысить индекс вязкости с 36 до 85—110, снизить содержание серы с 2 % до 0,04—0,1 %, почти на порядок уменьшить коксуемость, снизить температуру застывания. Подбирая условия (температуру, объемную скорость подачи сырья, катализатор), можно получать масла с высоким индексом вязкости практически из любых нефтей. Для ограничения деструктивных процессов и увеличения выхода целевых продуктов процесс часто осуществляют в две стадии. На первой стадии (температура 420—440 °С и давление 20—30 МПа) на АНМ-катализаторе происходит гидроочистка и гидрирование полициклических соединений. Высокое давление необходимо для глубокого расщепления и гидрирования полициклических аренов и циклоалканов, а также смол, вследствие чего снижается коксуемость и возрастает индекс вязкости.
На второй стадии (температура 320—350°С и давление 17—18 МПа) на бифункциональных катализаторах происходит доочистка масла и гидроизомеризация я-алканов. Так как изоалканы застывают при значительно более низкой температуре, чем алканы нормального строения, гидроизомеризация понижает температуру застывания масляных фракций и исключает операцию депарафинизации растворителями.
Гидроизомеризация керосино-газойлевых фракций на бифункциональных алюмоплатиновых катализаторах или сульфидах никеля и вольфрама на оксиде алюминия позволяет получать низкозастывающее дизельное топливо с температурой застывания до —35 °С.
Селективный гидрокрекинг. Процесс предназначен для удаления из сырья алканов нормального строения путем их гидрогенолиза с целью получения низкозастывающих топлив и масел. В качестве катализатора в этом процессе используют геометрически селективные цеолиты, размер входных окон которых (0,5—0,55 нм) позволяет свободно проходить в полости и реагировать там только молекулам нормальных алканов, имеющим диаметр 0,49 нм. Молекулы других углеводородов имеют больший диаметр (например, 2-метилпентан — 0,56; бензол— 0,58, циклогексан — 0,61 нм) и в полость цеолита попасть не могут. Для гидрирования образующихся продуктов превращения «-алканов в цеолит вводят обычные гидрирующие компоненты (металлы платиновой группы, никель, а также оксиды и сульфиды молибдена и вольфрама).
Селективному крекингу подвергают прямогонный бензин, а также бензин риформинга и бензин-рафинат после экстракции аренов с целью получения высокооктановых бензинов за счет снижения молекулярной массы и частичной изомеризации н-алканов. Комбинированный процесс, сочетающий риформинг и селективный гидрокрекинг, получил название селектоформин-га. Процесс заключается в переработке риформата или рафи-ната (после извлечения аренов) на катализаторе селективного гидрокрекинга при следующих условиях: температура около 360 °С, давление 3 МПа, объемная скорость 1 ч-1,. кратность циркуляции водородсодержащего газа 1000 м3/м3 сырья. В результате процесса октановое число бензина возрастает на 10— 15 пунктов. Селектоформинг эффективен на нефтеперерабатывающих заводах с большой мощностью по производству индивидуальных аренов.
Селективным удалением н-алканов из керосина и дизельного топлива получают низкозастывающее реактивное топливо (с температурой застывания от —50 до —60 °С) и зимнее дизельное топливо. Одновременно образуется фракция углеводородов С3—С5, используемая в качестве нефтехимического сырья. Процесс осуществляют по одно- или двухступенчатой схеме при температуре 350—400 °С, давлении 3—14 МПа, объемной скорости подачи сырья 0,5—5,0 ч-1, циркуляции водородсодержащего газа 500—1500 м3/м3 сырья. Выход реактивного топлива 75—90 %.
Селективный гидрокрекинг, так же как и гидроизомеризацию, используют для производства низкозастывающих масел из прямогонных фракций и рафинатов. Процесс проводят при температуре 300—430 °С, давлении 2—10 МПа, объемной скорости подачи сырья 0,5—2 ч-1. Выход масел составляет 80—87 %. Качество масел близко к качеству масел, получаемых низкотемпературной депарафинизацией растворителями. Температура застывания масел может быть понижена с +6°С до
— (40—50) °С. Для селективного гидрокрекинга масел у нас в стране разработан катализатор СГК-1 на основе высококремнеземного цеолита ЦВМ.
Гидродеароматизация. Каталитическая гидродеароматизация — основной процесс получения высококачественных реактивных топлив (керосина) из прямогонного и вторичного сырья. Содержание аренов в прямогонных среднедистиллятных фракциях в зависимости от происхождения нефти составляет 14—35%, в дистиллятах вторичного происхождения — до 70%. Переработку прямогонного сырья, содержащего серы меньше
0,2% и азота меньше 0,001 %, производят в одну стадию на платиноцеолитсодержащем катализаторе при температуре 280— 340 °С, давлении 4 МПа, объемной скорости подачи сырья 4 ч-1. Полнота удаления аренов при этом составляет 75—90 %. При большем содержании гетероатомов требуется предварительная гидроочистка. Вторичное сырье перерабатывается в более жестких условиях — при температуре 350—400 0С под давлением 25—35 МПа.
Несмотря на то, что гидрокрекинг является относительно дорогостоящим процессом, что связано с большими капитальными затратами на оборудование высокого давления и потреблением значительного количества водорода, он получил широкое промышленное развитие. Основные преимущества гидрокрекинга по сравнению с другими процессами переработки нефтяных фракций следующие:
гибкость процесса, т. е. возможность получения из одного сырья различных целевых продуктов, а также возможность переработки самых разных видов сырья — от тяжелых бензинов до нефтяных остатков;
большой пыход светлых продуктов; например, выход реактивного топлива можно увеличить с 2—3% на нефть до 15%, а выход зимнего дизельного топлива с 10—15 % до 100%'.
высокое качество получаемых продуктов.
ОЧИСТКА НЕФТЕПРОДУКТОВ
15.1. НАЗНАЧЕНИЕ И МЕТОДЫ ОЧИСТКИ
Получаемые в различных процессах переработки нефти фракции в большинстве случаев не являются готовыми товарными продуктами. Они содержат всевозможные примеси, присутствие которых делает эти фракции некондиционными, непригодными для использования. Для удаления нежелательных примесей нефтепродукты подвергают очистке.
Ниже изложены цели и методы очистки нефтепродуктов, зависящие от природы нефтепродукта и направлений его дальнейшего применения.
1. Дистилляты первичной перегонки некоторых нефтей содержат нафтеновые кислоты и другие кислые соединения, вредное влияние которых охарактеризовано в гл. И. Удаление этих соединений производят щелочной очисткой.
2. Коррозионноактивные сернистые соединения содержатся во всех фракциях, выделяемых при переработке сернистых нефтей. Особенно высока коррозионная способность сероводорода и низших меркаптанов. Для очистки газов и жидких, нефтепродуктов от серосодержащих соединений применяют- различные методы. Г азы, содержащие в основном сероводород и низшие меркаптаны, очищают с помощью щелочи, различных поглотителей, солей, адсорбентов. Для очистки жидких фракций от сероводорода и меркаптанов применяют щелочной метод и различные виды окислительной демеркаптанизации. Удаление более сложных серосодержащих соединений — тиофенов, сульфидов, дисульфидов, высших меркаптанов — производят гидроге-низационной очисткой (см. гл. 14).
3. Для получения топлив и масел с низкой температурой застывания применяют процесс депарафинизации, с помощью которого из средних дистиллятов удаляют жидкие парафины, а из масляных фракций — твердые углеводороды. Под твердыми углеводородами подразумевают все углеводороды, имеющие при комнатной температуре кристаллическое строение; они представляют собой многокомпонентную смесь алканов (от Ci6 и выше), йафтенов с длинными боковыми цепями нормального и изостроения, а также некоторого количества ароматических и нафтено-ароматических углеводородов. Существуют следующие методы депарафинизации: кристаллизация твердых углеводородов при пониженной температуре в отсутствие или в присутствии растворителей; карбамидная денарафинияация, использующая свойство карбамида (мочевины) образовывать с алканами твердые нерастворимые комплексные соединения; адсорбционная депарафииизация с применением цеолитов, селективно извлекающих из нефтяных фракций нормальные алканы.
4. При получении бензинов-растворителей, жидких парафинов, специальных масел и осветительных керосинов необходима очистка от ароматических соединений. Удаление аренов проводят сернокислотным методом.
5. Крекинг-бензины нуждаются в очистке не только от серосодержащих соединений, но и от алкадиенов и непредельных циклических соединений, которые легко полимеризуются с образованием смол. Для очистки от непредельных соединений применяют серную кислоту, различные катализаторы и адсорбенты.
6. При получении высококачественных масел из нефтяных фракций используют комплекс различных методов очистки. Из фракций последовательно удаляют асфальто-смолистые вещества, полициклические углеводороды с повышенной коксуемостью, смолистые вещества, парафины, серосодержащие и непредельные соединения. Для очистки применяют экстракционные, адсорбционные, гидрогенизационные методы.
15.2. ХИМИЧЕСКИЕ МЕТОДЫ ОЧИСТКИ
Очистка серной кислотой. Применение сернокислотного метода очистки сопровождается значительными потерями продуктов, подвергающихся полимеризации или растворяющихся в кислоте, а также образованием трудноутилизируемых отходов— кислых гудронов. Поэтому ведется поиск новых методов очистки, которые позволят отказаться от сернокислотного способа.
Реакции, происходящие при сернокислотной очистке. Алканы и циклоалканы при нормальной температуре не взаимодействуют с серной кислотой. Дымящая серная кислота при длительном контакте и тщательном перемешивании поглощает небольшие количества алканов.
Арены не вступают в реакцию с серной кислотой относительно невысокой концентрации. Концентрированная серная кислота, взятая в избытке, и олеум взаимодействуют с аренами. При этом образуются сульфокислоты и сульфоны, растворимые в серной кислоте:
C8He + H2S04 —> CeHsSOaOH + Н*0,
CeH5S020H -|- CeHg ——>• СиНбЗОгСвНв -|- НгО.
С алкенами серная кислота вступает в реакции присоединения. Легче всего взаимодействует кислота с алкенами, содержащими третичный углеродный атом.
При взаимодействии с алкенами образуются продукты при-
соединения двух типов: кислые эфиры (алкилсерные кислоты, моноалкилсульфаты) и средние эфиры (диалкилсульфаты):
RCH2 = СН2 + HOSO3H —* rch2ch2oso3h,
2rch2 = ch2 +HOSO3H —> rch2ch2oso2och2ch2r.
Кислые эфиры получаются при относительно низких темпе-, ратурах; они имеют кислотный характер, растворяются в воде, при нейтрализации щелочью дают соответствующие соли.
Полученные при сернокислотной очистке нефтяных фракций кислые эфиры концентрируются в кислом гудроне, а остатки этих эфиров из очищенного продукта удаляются дополнительной промывкой.
Средние эфиры образуются при повышенных (более 40 °С) температурах и при нагревании кислых эфиров. Средние эфиры нерастворимы в воде, но хорошо растворяются в углеводородах и органических растворителях.
Образование средних эфиров при сернокислотной очистке — нежелательное явление. Чтобы предотвратить его, сернокислотную очистку осуществляют при пониженной температуре.
Побочные реакции углеводородов. Наряду с основными реакциями углеводороды в присутствии серной кислоты вступают в побочные реакции, которые снижают эффективность очистки: алкилирование аренов алкенами, полимеризация, гидродегидрополимеризация (эту реакцию иногда называют сопряженной полимеризацией).
Образование алкилароматических углеводородов (алкилирование) является результатом взаимодействия кислых эфиров серной кислоты с ароматическими соединениями:
СвНв + RCH20S03H —>- CeH6CH2R + H2S04.
При полимеризации алкены уплотняются с образованием димеров, тримеров и тетр'амеров, которые растворяются в очищенном продукте, ухудшая его цвет.
Реакции алкенов с серной кислотой протекают, как правило, по карбкатионному механизму (см. гл. 11).
Реакции серосодержащих соединений. Сероводород окисляется с образованием элементарной серы и сернистого ангидрида:
H2S -f- H2S04 —> S -|- H2SO3 -I- H20 H2SCb —SO2 Ч- н2о h2s + h2so4 - > S + S02 + 2H20
Сера растворяется » очищаемом продукте и затем может «ступать в реакцию с углеводородами, вновь образуя сероводород. Поэтому перед кислотной очисткой сероводород из очищаемого продукта следует удалить.
Реакция меркаптанов с серной кислотой протекает в три стадии; продуктами реакции являются дисульфиды, которые легко растворяются в серной кислоте, и сернистый ангидрид:
RS
rsh + h2so4 —> ^>S02 + H20
ОН
RS RS
RSH \S02 —>¦ yS02 H2O
OH RS
RS
^>S02 —^ RSSR + SOj
_RS__
2RSH + H2S04 —> RSSR + SOs + 2H20.
При действии концентрированной серной кислоты на тиофен образуются тиофенсульфокислоты и оксид серы.
Дисульфиды, сульфиды, тетрагидротиофены и сульфоны в реакции с серной кислотой не вступают, но хорошо растворяются в ней, особенно при низких температурах.
Прочие реакции серной кислоты с компонентами нефтяных фракций. Имеющиеся в составе нефти азотсодержащие соединения взаимодействуют с серной кислотой, образуя сульфаты, переходящие в кислый гудрон. Нафтеновые кислоты частично растворяются в серной кислоте, а частично сульфируются, причем карбоксильная группа нафтеновых кислот при сульфировании не разрушается. Продукты взаимодействия нафтеновых и серной кислот ослабляют эффективность действия серной кислоты на другие соединения, поэтому целесообразно перед сернокислотной очисткой предварительно удалить из очищаемого продукта нафтеновые кислоты.
В зависимости от того, для какой цели применяют сернокислотную очистку, подбирают концентрацию кислоты и технологический режим процесса. При очистке, целью которой является удаление смолистых веществ из смазочных масел и повышение качества осветительных керосинов, применяют 93%-ю кислоту. Для деароматизации используют 98%-ю кислоту или олеум. Легкую очистку бензина, предназначенную для улучшения цвета, проводят серной кислотой концентрацией 85 % и ни_ же. Применение разбавленной кислоты там, где это нозможно, предпочтительнее, так как кислый гудрон образуется н меньших количествах, ослабляются процессы полимеризации.
Сернокислотную очистку большинства фрикций осуществляют, не прибегая к предварительному подогреву, поскольку повышение температуры способствует полимеризации алкенов.
Однако в некоторых случаях приходится повышать температуру. Так, при 50—85 °С проводят деароматизацию бензинов-растворителей, осветительных керосинов, парфюмерных и медицинских масел.
Повышение температуры способствует полимеризации непредельных углеводородов, поэтому сернокислотную очистку большинства фракций проводят без подогрева очищаемого сырья. При деароматизации нефтяных фракций (бензинов-раст-ворителей, осветительных керосинов, медицинских и парфюмерных масел) температура очистки повышается. При повышенной температуре осуществляют сернокислотную очистку смазочных масел, поскольку подогрев позволяет снизить вязкость сырья, улучшить условия разделения очищенного продукта и кислого гудрона.
Выбор времени контакта сырья и серной кислоты определяется рядом факторов. Длительный контакт нефтепродукта с кислым гудроном ухудшает цвет и стабильность очищенного продукта, а при слишком малом времени контакта не полностью используется кислота. Важное значение имеют степень диспергирования кислоты в продукте и требуемое время отстоя кислого гудрона.
Для сернокислотной очистки используют установки периодического и непрерывного действия.
Очистка щелочью. Щелочная очистка (защелачивание) предназначена для удаления из нефтепродуктов кислых и серосодержащих соединений: нафтеновых и жирных кислот, а также фенолов, переходящих в дистилляты из нефти или образовавшихся в процессах вторичной переработки; кислот, образовавшихся в продукте после его сернокислотной очистки; сероводорода и низших меркаптанов. С другими компонентами нефтепродуктов щелочь не реагирует.
Реакции, происходящие при щелочной очистке. Свободные кислоты, находящиеся в дистилляте, вступают в реакцию со щелочью, образуя соли, которые в основном сосредоточены в щелочном растворе:
RCOOH + NaOH —•> RCOONa + Н20.
Фенол взаимодействует со щелочью с образованием фенолятов:
CeH5OH + NaOH -—>¦ CeH5ONa + Н20.
Средние эфиры серной кислоты под действием щелочи омы-ляются, превращаясь в соответствующие соли, также переходящие в щелочной раствор:
(СгНеОЬЯОг + 2NaOII - * 2С»Н6ОН + Na2SO„.
Часть солей задерживается и нефтепродукте, для их удаления обработанный щелочью дистиллят промывают водой.
Реакция нейтрализации щелочью нафтеновых кислот и фенолов имеет обратимый характер. Нафтенаты и феноляты в присутствии воды гидролизуются, образуя исходные продукты. Степень гидролиза зависит от условий процесса: увеличивается с повышением температуры и понижается с ростом концентрации раствора щелочи. Поэтому очистку целесообразно проводить при невысоких температурах, используя концентрированные растворы. Однако в этих оптимальных условиях нейтрализации образуются стойкие эмульсии типа «кислое масло в водной щелочи», которые имеют в качестве внешней (непрерывной) фазы воду и называются гидрофильными.
Возникновению эмульсий способствуют сами продукты нейтрализации— натриевые соли нафтеновых и сульфокислот. Чтобы предотвратить образование эмульсий, щелочную очистку масел проводят низкоконцентрированными щелочными растворами при повышенных температурах.
Сероводород реагирует со щелочью с образованием кислых и средних солей:
H2S + NaOH —> NaHS + H20,
H2S + 2NaOH —> Na2S + 2H20,
Na2S + H2S —> 2NaHS.
Сульфид натрия получают при избытке щелочи, а кислую соль — при недостатке.
Меркаптаны, взаимодействуя со щелочью, образуют мер-каптиды:
RSH + NaOH RSNa + Н20.
Удаление меркаптанов щелочной промывкой связано с большими трудностями. Кислые свойства меркаптанов снижаются при увеличении длины углеводородной цепи, и вследствие этого высшие меркаптаны не реагируют со щелочью. Помимо реакций образования меркаптидов в присутствии кислорода воздуха происходит окисление меркаптанов с получением дисульфидов:
2RSH + Ог —> RSSR + H2O2 2RSH + H2O2 —* RSSR + 2H2O.
4RSH + 02 —> 2RSSR + 2H20,
Дисульфиды в воде нерастворимы и переходят в очищаемый дистиллят, еще больше снижая тем самым эффект извлечения меркаптанов.
Очистка поглотительными растворами. Для очистки газов от сероводорода широко применяют поглотительные растворы. При низких температурах сероводород поглощается растворами, а при повышенных температурах или при продувке воздухом происходит регенерация поглотительного раствора и десорбция сероводорода. Наибольшее распространение получили этаноламиновый, фенолятный и фосфатный методы, в основе которых лежат следующие обратимые реакции:
2NH2 (СН2СН2ОН) + H2S (CH2CH2OHNH3)2S,
моноэтаноламнн
CeH6ONa + H2S СвНбОН + NaHS,
фенолят натрия
k3po4 + h2s k2hpo4 + khs.
трикалийфосфат
Этаноламиновые растворы наряду с сероводородом поглощают диоксид углерода:
NH2 (СН2СН2ОН) + С02 + Н20 (CH2CH2OHNH3) НС03.
В последнее время растворы моноэтаноламина применяют также для очистки от сероводорода сжиженных газов.
Для избирательной очистки от сероводорода газов, содержащих оксид и диоксид углерода, используют мышьяково-содо-вый метод.
15.3. АДСОРБЦИОННЫЕ И КАТАЛИТИЧЕСКИЕ МЕТОДЫ ОЧИСТКИ
В последние годы разработаны методы, позволяющие вовлекать в переработку на установках каталитического крекинга не только дистиллятное, но и тяжелое сырье (вакуумный газойль с концом кипения 560 °С) после его облагораживания методом гидроочистки.
Переработка остаточного сырья является более сложной задачей, которая решается путем использования металлостойких катализаторов и специальных добавок — пассиваторов ванадия, никеля, железа — и комбинированием процесса каталитического крекинга с процессами подготовки и облагораживания сырья. Весьма перспективно облагораживание остаточного сырья в процессе адсорбционно-каталитической очистки (АКО) от асфальтенов, тяжелых металлов и частично серы и азота на циркулирующем мелкодисперсном адсорбенте. В процессе достигается глубина удаления тяжелых металлов и асфальтенов на 89—95 %, серы на 35—40 %, азота на 50—60 %, коксуемость продукта снижается на 75—80 %- Широкая газойлевая фракция адсорбционно-каталитической очистки (АКО) характеризуется повышенным содержанием непредельных соединений, тяжелых ароматических углеводородов, смол, металлов, вследствие чего нуждается н гидрооблагораживании перед тем, как будет использоваться в процессе каталитического крекинга.
Процесс гидрооблагораживания осуществляется по двухстадийной схеме при давлении 7,5 МПа на системе из нескольких катализаторов.
Материальный баланс переработки мазута процессами АКО — гидрооблагораживание — каталитический крекинг
Поступило, %(масс.): мазут водород
100
1,6
4.43
Получено, %(масс.): сухой газ
Адсорбционная очистка. При переработке нефти широко используют способность некоторых естественных глин, синтетических алюмосиликатов, силикагеля, алюмогеля и других веществ адсорбировать на своей поверхности различные компоненты и примеси. Упомянутые вещества являются полярными адсорбентами, их молекулы состоят в основном из оксидов кремния и алюминия. Физико-химические основы процесса адсорбции освещены в гл. 5.
Адсорбенты служат для очистки масляных фракций от нежелательных компонентов; доочистки предварительно обработанных селективными растворителями и депарафинированных масляных фракций; доочистки жидких и твердых парафинов; очистки индивидуальных аренов; осушки углеводородных газов и нефтяных фракций; для выделения из жидких фракций нормальных алканов.
При адсорбционной очистке от нежелательных компонентов из очищаемых масляных фракций удаляются смолы к поли-циклические ароматические компоненты. Очистку проводят в аппаратах колонного типа при противоточном движении продуктов— адсорбент движется сверху вниз, а носитель (масляная фракция, подвергающаяся очистке)—снизу вверх. В качестве адсорбента используют синтетический алюмосиликат с зернами размером 0,25—0,50 мм. Адсорбционная очистка обеспечивает более высокий выход масла, чем селективная, поскольку при адсорбции удаляются только нежелательные компоненты и полностью сохраняются ценные углеводороды исходного сырья. Масла, полученные адсорбционной очисткой, обладают высокой стабильностью против окисления. Широкому внедрению процесса препятствуют высокие эксплуатационные затраты, а также трудности в конструктивном исполнении установок. Процесс применяют для получения трансформаторного масла и высокоароматизированного масла — теплоносителя.
Адсорбционная доочистка масляных фракций, прошедших несколько ступеней очистки, служит для удаления из очищенных фракций всевозможных примесей — кислого гудрона, солей нафтеновых кислот, избирательных растворителей, смол. Существуют два метода адсорбционной очистки — контактная очистка и перколяция.
При контактной очистке масло смешивают с адсорбентом, смесь нагревают и выдерживают при определенной температуре, затем масло отфильтровывают. Нагрев необходим, чтобы понизить вязкость масла и облегчить его проникновение во внутренние поры адсорбента. В качестве адсорбента применяют природные глины (отбеливающие земли)—гумбрин, бентониты, зикеевскую и балашеевскую опоки, а также синтетические алюмосиликаты тонкого помола. Недостатки контактной очистки: значительная потеря масла с отработавшими глинами, низкая активность и трудная регенерируем ость глин.
Перколяция представляет собой периодический процесс — фильтрование масла через неподвижный слой зерненого адсорбента. Адсорбент — отбеливающие земли с размером зерен
0,3—2,0 мм.
Адсорбционная доочистка твердых алканов служит для удаления нестабильных, красящих и обладающих запахом веществ; проводят ее теми же методами, которые используют для доочистки масел (контактная и перколяционная доочистка). Из жидких парафинов посредством адсорбционной доочистки можно удалять ароматические и серосодержащие соединения, а также смолистые вещества.
Каталитическая очистка. Для повышения качества нефтепродуктов, полученных при первичной перегонке и вторичных процессах, применяют каталитическую очистку. В промышленной практике распространены следующие методы очистки в присутствии катализаторов: а) очистка от сернистых соединений под давлением водорода в присутствии алюмокобальт-молибденоных или алюмоникельмолибдеповых катализаторов (гидроочистка); б) очистка от непредельных углеводородов с помощью алюмосиликатов; в) очистка от сернистых соединений с помощью природных бокситов и алюмосиликатных катализаторов; г) каталитическая демеркаптанмзация (процесс Мороке).
Процессы гидрогенизадионной очистки подробно рассмотрены в гл. 14.
Каталитической очистке от непредельных углеводородов подвергают обычно бензины, полученные каталитическим крекингом, пропуская пары бензина через слой алюмосиликатного катализатора.
Широко распространен процесс каталитической демеркап-танизадии сжиженных газов и нефтяных фракций. Меркаптаны превращаются в нейтральные дисульфидные соединения путем окисления воздухом на специальном катализаторе в щелочной среде:
4RSH + 02 —> 2RSSR + 2H20.
15.4. МЕТОДЫ ОЧИСТКИ
С ПРИМЕНЕНИЕМ ИЗБИРАТЕЛЬНЫХ РАСТВОРИТЕЛЕЙ
Очистку с помощью избирательных растворителей наиболее широко применяют в производстве масел. Современная технология получения масел из нефтей восточных районов нашей страны включает несколько процессов очистки с применением селективных растворителей: удаление смолисто-асфальтеновых веществ деасфальтизацией гудрона; выделение полициклических ароматических углеводородов с короткими цепями и смолистых соединений при так называемой селективной очистке масел; извлечение твердых алканов (депарафииизация).
Особую группу составляют процессы извлечения аренов из продуктов, полученных каталитическим риформированием или пиролизом нефтяных фракций.
При селективной очистке масел растворители хорошо растворяют нежелательные компоненты, не затрагивая совсем или растворяя в незначительной степени те соединения, которые нужно сохранить в составе масел. При депарафинизации и деасфальтизации растворители, наоборот, хорошо растворяют желательные компоненты, вредные примеси осаждаются из раствора.
В качестве селективных растворителей используют различные органические соединения: спирты, альдегиды, кетоны, амины, иитросоединения, простые и сложные эфиры. В промышленности применяют при деасфальтизации пропан, при селективной очистке — жидкий сернистый ангидрид, нитробензол, фенол, фурфурол, крезол, при депарафинизации—смесь кето-на (ацетона или метилэтилкетона) с бензолом и толуолом, пропан, дихлорэтан, карбамид, при извлечении аренов — ди-, три-, тетраэтиленгликоли, сульфолан, пропиленкарбонат, N-ме-тилпирролидон и др.
Ниже перечислены общие требования, предъявляемые к растворителям:
1. Растворитель должен обладать ярковыраженной избирательной растворимостью в широком интервале температур.
2. Для облегчения процесса разделения фаз разница между плотностями растворителя и сырья должна быть значительной.
3. Для улучшения условий регенерации растворителя его температура кипения должна быть значительно ниже температуры кипения сырья.
4. Для снижения энергетических затрат желательно, чтобы растворитель имел низкую теплоту испарения.
На растворяющую способность растворителя оказывают влияние его полярность и строение углеводородного радикала при функциональной группе. Строением углеводородного радикала определяют дисперсионные межмолекулярцые взаимодействия при растворении.
В процессах очистки масел избирательными растворителями важную роль играют такие показатели, как критическая температура растворения масляной фракции в растворителе (КТР), температура процесса, соотношение растворителя и сырья. Если в качестве растворителя применяют сжиженные газы (пропан, сернистый ангидрид), большое значение имеет давление.
При смешении избирательного растворителя с нефтяной фракцией в сырье первоначально растворяется лишь небольшое количество растворителя. При увеличении соотношения растворитель : сырье появляется двухфазная система. Одна из фаз включает сырье с небольшой примесью растворителя, другая — растворитель, содержащий извлеченные из сырья вещества. Если и дальше увеличивать кратность подачи растворителя, то наступит момент, когда растворитель полностью смешается с фракцией.
Повышение температуры при данной кратности растворителя также увеличивает растворимость углеводородов нефтяной фракции до тех пор, пока не будет достигнута КТР, выше которой углеводороды полностью смешиваются с растворителем и система становится однофазной.
Н. И. Черножуков и Ю. А. Пинкевич установили следующие закономерности, связанные с КТР избирательных растворителей и нефтепродуктов: а) чем больше в очищаемом дистилляте содержится ароматических непредельных углеводородов, тем ниже его КТР; б) чем выше температура . кипения дистиллята из одной и той же нефти, тем выше его КТР;
в) очищенный определенным растворителем продукт (рафн-нат) имеет более высокую КТР, чем сырье; г) чем глубже очистка, тем выше разница между КТР очищенной и неочищенной фракции.
Очистку следует проводить при температуре, не превышающей КТР, т. е. в условиях, когда существует двухфазная система. Выбор конкретной температуры зависит от требований, предъявляемых к качеству очищенного продукта, и необходимого количества отбираемого материала. Для различных растворителей и сырья температуру очистки находят опытным путем.
Количество подаваемого для очистки растворителя зависит от его свойств, состава исходного сырья, требуемой степени очистки, температуры и применяемого способа экстракции. Увеличение кратности подачи растворителя приводит к уменьшению выхода рафината и улучшению его качества. Так, при обработке фенолом одного и того же образца масла были получены следующие результаты:
| Кратность |
Выход | Индекс |
Коксуемо! |
|
рафнната, % | вязкости |
% | |
| 3: 1 |
66,3 | 87,5 | 1,0 |
| 6 : 1 | 50,0 |
92,8 | 0,8 |
| 12 : 1 | 34,0 | 97,5 |
0,6 |
Методы экстракции подразделяют на однократный, многократный периодический и противоточный. При однократном методе нефтепродукт смешивают со всем количеством растворителя, а затем направляют смесь на разделение; из образовавшихся рафинатной и экстрактной фаз отгоняют растворитель. При многократном периодическом способе исходное сырье обрабатывают отдельными порциями растворителя, добавляя каждую последующую порцию после отделения экстрактной фазы. Степень очистки при многократном методе выше, чем при однократном.
Еще выше эффективность противоточного метода, при котором очищаемый продукт непрерывно движется навстречу растворителю. Сырье по мере контакта с растворителем все в большей степени освобождается от нежелательных компонентов, его КТР повышается, и, следовательно,- для донзвлечеНия остающихся в рафинате нежелательных компонентов требуется более высокая температура. Поэтому на входе в экстрактор температура'растворителя должна быть выше, чем у очищаемого сырья. Разницу между температурами растворителя и сырья называют температурным градиентом экстракции.
Деасфальтизация гудрона. На промышленных установках для пзилечепии смолисто-асфальтепоиых пещестп из остатка вакуумной перегонки нефти — гудрона — и основном применяют жидкий пропан, который при температурах, близких к критической (96,8 °С), не растворяет смолы и асфальтены, выпадающие в осадок. Объясняется это тем, что с приближением температуры пропана к критической резко снижается его плотность и увеличивается молярный объем, в то время как эти показатели высокомолекулярных углеводородов изменяются незначительно. В итоге уменьшаются силы притяжения между молекулами растворителя и углеводородов, что приводит к выпадению смолисто-асфальтеновых веществ в осадок.
Деасфальтизадию проводят в экстракционных колоннах и роторно-дисковых контакторах (РДК) под давлением, превышающим давление насыщенных паров сжиженного пропана. Из верхней части колонны или РДК выводят раствор деасфаль-тизата — основное количество пропана с растворенными в нем масляными фракциями, из нижней — раствор асфальта в пропане.
Повышение температуры в верхней части колонны или РДК позволяет получить более светлый деасфальтизат с меньшей коксуемостью. Однако выход деасфальтизата уменьшается, так как с приближением температуры к критической начинается переход в асфальт не только смол и асфальтенов, но и высокомолекулярных аренов полициклического строения. При достижении критической температуры растворителя все углеводороды выделяются из раствора. Уменьшение температуры повышает растворяющую способность пропана, в растворе удерживаются не только алкано-циклоалканы и высокоиндексные арены, но и смолисто-асфальтеновые соединения.
При некоторой определенной температуре увеличение подачи пропана сначала приводит к улучшению осаждения из сырья смолисто-асфальтеновых соединений. Однако при избытке пропана смолы начинают растворяться в нем и переходят в деасфальтизат, повышая его вязкость и коксуемость. Объемное соотношение пропан: сырье составляет от 4:1 до 10:1, причем для малосмолистых нефтей необходимо поддерживать более высокое соотношение.
Селективная очистка. В результате селективной очистки вакуумных дистиллятов и деасфальтизированных гудронов повышается индекс вязкости масел, снижаются содержание сернистых соединений и коксуемость, улучшаются цвет и вязкостно-температурные свойства. При селективной очистке образуются рафинатный раствор, содержащий в основном целевые алканоциклоалканы, и экстрактный раствор, в котором концентрируются смолы и арены.
Для эффективного проведения очистки иажно обеспечить равномерное подрастание температуры от нижней части экстрактора к нерхнсй, что позволяет наиболее четко разделить желательные и нежелательные компоненты сырья. Увеличение соотношении растворитель : сырье позволяет улучшить качестно очищенного масла; при недостаточном разбавлении в рафинат переходит много тяжелых аренов и смол, ухудшаются цвет и индекс вязкости рафината.
В качестве растворителя ранее применяли сернистый ангидрид и нитробензол, а в настоящее время используют фенол, фурфурол, N-метилпирролидон.
Растворимость компонентов масел в полярных и неполярных растворителях изменяется при введении различных добавок. Так, при добавлении к фенолу воды повышается его селективность и уменьшаются растворяющие свойства. Такое действие воды может быть объяснено образованием водородных связей. Как показали исследования, добавка воды к фенолу (6—8%) повышает выход рафината и лишь в незначительной степени снижает индекс вязкости, что является показателем достаточной степени извлечения полициклических углеводородов и повышения селективности фенола.
Депарафинизация топлив и масел. Депарафинизация топлив и масел предназначена для снижения температуры застывания очищаемых продуктов. Удаленные при очистке продуктов жидкие и твердые парафины являются ценным химическим сырьем. Для депарафинизации топлив используют процессы карбамидной депарафинизации (см. гл. 6) и адсорбционного извлечения. При очистке масляных фракций наиболее широкое распространение получил метод кристаллизации с использованием растворителей.
Для полного извлечения из рафинатов селективной очистки твердых парафинов необходимо глубоко охладить сырье. Однако при охлаждении заметно увеличивается вязкость рафината, а это затрудняет рост кристаллов парафинов. Было установлено, что добавление растворителя позволяет, не повышая вязкости сырья, глубоко охладить его и тем самым обеспечить выделение парафинов.
Первой стадией кристаллизации является выделение из пересыщенного раствора зародышей кристаллов — мельчайших частиц кристаллизующегося вещества. Затем кристаллы растут, причем наиболее легко осуществляется рост на острых углах зародышей кристаллизации. Если количество зародышей кристаллов невелико, то в процессе кристаллизации образуются крупные кристаллы. Скорость роста кристаллов на центрах кристаллизации может быть определена по уравнению:
dx
гдс -jj---количество нещсства, никристалли.-юиаишегоси н единицу вре
мени; D — коэффициент диффузии молекул угленодорода в насыщенном растворе; 6 — средняя длина диффузионного пути; S — поверхность выделив-
Шейся твер1°й фазы; х — концентрация пересыщенного раствора; Xi — растворимость зародышей кристаллов при данной степени их дисперсности.
Коэффициент диффузии D находят по уравнению:
где R_универсальная газовая постоянная; Т — абсолютная температура кри
сталлизаций N — число Авогадро; г—-средний радиус молекулы твердого Углеводород’ 'П — динамическая вязкость среды.
Подст<вляя значение D в первое уравнение, можно преобразовать егс к следующему виду:
D ST
Следовательно, скорость выделения твердой фазы из раствора зависиг от вязкости среды, средней длины диффузионного пути, среднего радиуса молекул твердых углеводородов и разности ме»ДУ концентрацией раствора и растворимостью выделившейся при температуре Т твердой фазы.
На эффективность процессов депарафинизации влияют качество сырья> природа, состав и кратность подачи растворителя, добавляемого к сырью, скорость охлаждения раствора сырья.
Полно!3 выделения твердых углеводородов при депарафинизации зависит от четкости фракционирования масляных дистиллятов. Дистилляты широкого фракционного состава содержат молекулы твердых углеводородов, значительно различающихся по структуре, что приводит к образованию эвтектических смесей неД°развитых кристаллов отдельных групп углеводородов и к затруднению отделения твердой фазы от жидкой. Поэтому предпочтительнее подвергать депарафинизации узкие фракции. 'Увеличение вязкости масляных фракций с повышением температур выкипания затрудняет диффузию молекул твердых углеводородов к центрам кристаллизации. При этом появляются дополнительные центры кристаллизации, уменьшаются конечные размеры кристаллов, ухудшаются условия выделения твердых углеводородов. Поэтому выделение твердых углеводород08 непосредственным охлаждением масляных фракций возмо>*но только для маловязких парафинистых дистиллятов. В других случаях осуществляют депарафинизацию охлаждением в п рисутствии растворителей.
Раствор^ители, применяемые при депарафинизации, должны обладать следующими свойствами: 1) при температуре про
цесса не растворять твердые углеводороды сырья, а растворять жидкие; 2)) позволять поддерживать минимальную разность между тем пературами депарафинизации и застывания депара-финированного масла; разность между этими температурами носит название температурного эффекта депарафинизации (ТЭД); 3) иметь достаточно низкую температуру застывания, чтобы не кристаллизоваться при температуре депарафинизации; 4) быть коррозионно-неагрессивными.
В качестве растворителей при депарафинизации применяют неполярные вещества — пропан, узкую бензиновую фракцию (нафту) и полярные — ацетон, метилэтилкетон, дихлорэтан. Неполярные растворители полностью растворяют жидкую часть масла, а полярными растворителями она растворяется слабо. Твердые углеводороды также гораздо лучше растворяются неполярными растворителями. Чтобы повысить растворяющую способность полярных растворителей, к ним добавляют неполярные углеводороды; такие полярные растворители, как ацетон, метилэтилкетон, дихлорэтан используют в смеси с бензолом и толуолом или только с толуолом.
Выбор оптимальной скорости охлаждения зависит от фракционного состава сырья, типа растворителя и кратности подачи растворителя по отношению к сырью. Высокая скорость охлаждения (300°С/ч) способствует образованию большого числа центров кристаллизации и, как следствие, появлению мелких кристаллов, снижению выхода депарафинированного масла и скорости фильтрации, повышению содержания остаточных масел в твердой фазе. Скорость охлаждения играет особенно важную роль в начальный период охлаждения — в момент образования первичных центров кристаллизации. При дальнейшем охлаждении скорость охлаждения может быть увеличена.