Глава 12 термические превращения углеводородов нефти
Глава 12
ТЕРМИЧЕСКИЕ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ НЕФТИ
12.1. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ТЕРМИЧЕСКИХ ПРОЦЕССОВ
Термодинамика. Термодинамическую вероятность протска-яия химической реакции определяют величиной изменения а процессе свободной энергии Гиббса. Реакция протекает слева направо при отрицательном значении энергии Гиббса. Для всех углеводородов, кроме ацетилена, с повышением температуры энергия Гиббса возрастает. Чем большим запасом свободной энергии обладает молекула, тем менее она стабильна,, т. е. термодинамическая стабильность всех углеводородов (кроме ацетилена) с повышением температуры падает. Энергия Гиббса алканов и циклоалканов увеличивается быстро, алкенов и аренов — медленно. Вследствие этого соотношение термодинамической устойчивости углеводородов различных классов с температурой меняется: при температуре до 227 °С наиболее устойчивы алканы, при более высокой температуре — алкены, алкадиены, арены. Следовательно, для переработки алканов в алкены достаточно простого нагревания до высокой температуры. Однако алкены при любой температуре неустойчивы к вторичным реакциям, например к полимеризации. Кроме того, даже при относительно низкой температуре термодинамически возможен распад углеводородов на элементы. Поэтому общее термодинамическое равновесие системы со временем сдвигается в сторону глубоких превращений (с образованием водорода, метана, смолы, кокса), и время становится одним из основных параметров, определяющих состояние системы. Конечный состав продуктов высокотемпературных процессов в значительной степени определяется кинетическими закономерностями. Если целью процесса является получение максимального выхода алкенов, то реакцию надо остановить в момент их наибольшей концентрации и не дать возможности системе приблизиться к конечному термодинамическому равновесию.
Кинетика и механизм процесса. Термические реакции углеводородов протекают главным образом по радикально-цепному механизму.
Радикально-цепной механизм термических превращений алканов был-предложен американским химиком Ф. Райсом (1934 г.). Большое значение в создании теоретических основ высокотемпературных реакций углеводородов имела разработанная академиком Н. Н. Семеновым общая теория цепных: реакций (1958 г.).
Молекулярные реакции играют незначительную роль, а ионные реакции в условиях термических газофазных некаталитических процессов практически отсутствуют, так как гетеролити-ческий распад С—С-связи требует энергии да 1200 кДж/моль* в то время как гемолитический да360 кДж/моль. Большие затраты энергии при гстеролитпчсском процессе обусловлены необходимостью преодоления электростатического взаимодействия ионов и их нестабильностью в несольватированном состоянии.
Раднкально-цепной процесс термического разложения углеводородов, как любой цепной процесс, складывается и:* трех стадий: инициирование цепи, продолжение цепи, обрыв цепи.
Инициирование цепи. Распад алифатических углеводородов на радикалы осуществляется преимущественно по наиболее слабой связи С—С. Энергия С—Н-связей в алканах всегда выше энергии С—С-связи. Например, энергия, необходимая для разрыва С—С- и С—Н-связей в молекуле этана, равна соответственно 360 и 410 кДж/моль. Это означает, что при одинаковых предэкспоненциальных множителях отношение констант скоростей распада этана по связям С—С и С—Н составит при
600 °С приблизительно 1 • 103356, т. е. распад по связи С—Н несу
ществен относительно распада по связи С—С.
В нормальных алканах с длинной цепью энергия разрыва С—С- и С—Н-связей несколько уменьшается к середине цепи, однако первая всегда остается значительно меньше второй:
СНз - СН2 - СН2 — СН2 - СН2 - СН2 - СН2 - СНз.
Энергия связей, кДж/моль:
С—С 335 322 314 310 314 322 335
С—Н 394 373 364 360 360 364 373 394
С повышением температуры различие в прочности связей уменьшается, поэтому при умеренной температуре (400—500 °С) разрыв углеводородной цепи происходит посредине, а при более высокой температуре разрыв возможен и по другим связям.
Связи С—С в циклоалканах несколько менее прочны, чем в нормальных алканах: в циклогексане на 8 кДж/моль, в цик-лопентане на 25 кДж/моль.
Связи С—С и С—Н в алкенах у атома углерода с двойной связью значительно прочнее, а в Р-положении— сильно ослаблены по сравнению с алканами (цифры — энергия связи в кДж/моль):
Энергия раскрытия я-связи в алкене при сохранении о-связи равна 249 кДж/моль:
СН2 = СН2 —> СН2 — СН2 — 249 кДж/моль.
Если двойная связь является сопряженной, то энергия раскрытия я-связи примерно на 50 кДж/моль меньше:
СН2 == СН — СН = СН2 - » СН2 = СН-СН-СН2-
— 188 кДж/моль.
В аренах связи С—Н и С—С прочнее, чем в алканах, а связи, сопряженные с ароматическим кольцом, ослаблены. Сопряжение с кольцом снижает прочность связи примерно в той же мере, как и сопряжение с двойной связью (цифры — энергия связи в кДж/моль):
Н
325
Легкость гемолитического разрыва связи в углеводороде значительно зависит также от устойчивости образующихся радикалов. Энергия диссоциации молекулы на радикалы отличается от энергии связи на величину энергии сопряжения неспаренного электрона с другими связями в молекуле. Так, энергия диссоциации связи Салиф—Салиф в гексафенилэтане равна всего «42 кДж/моль:
-]зС~С(-\У);з 2^y;-J3C.-42 кДж/моль.
При гомолитическом распаде связи происходит переход электронов двухэлектронной связи на орбитали разных атомов, при этом образуются два радикала или бирадикал:
СН3 СН3 -^ 2СН3
.СН2 Н2С' ’* СН2
^ СН2СН2СН2СН2СНгСН2
Радикалы могут образовываться и по бимолекулярной реакции:
СзНв + С2Н4 —>¦ С2Н5 -j- С3Н5 ~¦ 155 кДж/моль;
СгНв Ч- С2Н4 ¦—> 2С2Н5.
Роль бимолекулярного процесса по сравнению с мономоле-кулярным возрастает с повышением давления и снижением температуры.
Продолжение цепи (реакции радикалов). Радикалы представляют собой химически ненасыщенные частицы и обладают высокой реакционной способностью. Стабильность
(СвН5)з С > (CeHsb сн > сн2 = снсн2 > СбН5СН2 >
> (СНз)зС > с8н5>
!СНзСН2СНСНз 'I СН3СН2СН2 >>сн2 = сн.
СНз J
Реакционная способность радикалов в такой же последовательности возрастает.
Различают следующие реакции радикалов.
1. Замещение (отрыв атома водорода):
R + R'H —* RH + R' ±Q.
По правилу Поляни—Семенова энергия активации Е; (в кДж/моль) реакций углеводородных радикалов с углеводородными молекулами связана с тепловым эффектом реакции Q соотношениями:
а
для экзотермических реакций Е& = 48—0.25Q; для эндотермических реакций Еа = 48 + 0.75Q.
Расчет энергии активации нескольких возможных реакций метального радикала с пропиленом позволил получить следующие результаты:
о в»
а»
кДж/моль кДж/моль
СН3 + СН2 = СН-СН3
Разница энергии активации в 25 кДж/моль при одинаковых предэкспоненциальных множителях соответствует отличию в константах скорости при 700 °С примерно в 20 раз, т. е. первая реакция будет протекать в 20 раз быстрее, чем вторая и третья.
Энергия активации реакций алкильных радикалов с алка-
нами R + CH3CH2CH2CH3—»- RH + CH3CH2CHCH3 составляет
40—50 кДж/моль.
Зная энергию активации реакций отрыва радикалом различных атомов водорода, молено определить константы скоростей реакций при заданной температуре и их соотношение. Умножив относительную скорость на число первичных, вторичных и третичных атомов водорода в молекуле, можно найти вероятность образования соответствующих радикалов. В случае н-бутаиа прн 600 "С соотношение вероятности образования иер-вичных и вторичных бутильных радикалов равно 3 : 4. Для простейших алканов расчетные данные практически совпадают с экспериментальными.
2. Распад радикалов с образованием ненасыщенных молекул и новых свободных радикалов меньшей молекулярной массы.
Распад протекает преимущественно по наиболее слабой связи, находящейся в P-положении относительно атома углерода с неспаренным электроном (Р-правило):
Н
|Р
СНз^-СН-СНТСНз
СНз—СН—сн2 { СНз — [сн3—сн—сн2]* + сн3
![]()
СНз—СН—СН—СНз -> [СНз—СН СН СНз^Н- Н
СН3—СН=СН—СНз
Реакции Р-распада углеводородов эндотермичны. Энергия активации распада крупных радикалов (Сз и выше) составляет
110—170 кДж/моль. Низшие радикалы СН3, С2Н5 устойчивы к распаду. Если распад неразветвленного вторичного алкильного радикала может произойти по нескольким направлениям, то энергетически более выгоден процесс, при котором образуется радикал с наибольшей молекулярной массой.
3. Присоединение радикала по кратной связи (реакция, обратная Р-распаду):
Присоединение радикалов по двойной связи протекает с выделением теплоты (76—105 кДж/моль). Энергия активации, рассчитанная по правилу Поляни—Семенова, составляет 22— 29 кДж/моль.
4.' Изомеризация свободных радикалов. Предполагают, что изомеризация протекает через циклическое переходное состояние:
:н-снасн3
С Из—CIIv-Civ СН2—CII3—СН;.—CH2 —
Кроме 1,5-изомеризации радикалов происходит также 1,4-, 1,6- и 1,7-изомеризация. Трех- и четырехчленные циклы слишком напряжены, поэтому 1,2- и 1,3-изомеризация алкильных радикалов практически неосуществима. Изомеризация алкильных радикалов протекает с небольшим положительным тепловым эффектом. Энергия активации процесса составляет 50— 90 кДж/моль.
Для аренов наблюдается 1,2-переход фенильного радикала:
Н2СЧ
Н2С
НС—)) -+ НС.
НзС НзС НзС
Обрыв цепи. Обрыв цепи осуществляется следующими реакциями.
1. Рекомбинация радикалов:
2С2Н5 —>¦ С4Н10
2. Диспропорционирование радикалов (процесс, обратный бимолекулярной реакции образования радикалов):
2С2Н5 —> СгНб -|- С2Н4
Энергия активации этих реакций равна нулю.
Кажущаяся энергия активации радикально-цепной реакции термического крекинга н-бутана составляет 245 кДж/моль.
Соотношение скоростей реакций радикалов. При температуре выше 280 °С скорость реакций (3-распада алкильных радикалов больше скорости замещения, вследствие чего длинные алкильные радикалы подвергаются крекингу.
Для реакций радикалов с алкенами возможна конкуренция мономолекулярного распада с бимолекулярными процессами присоединения и замещения. Направление и скорость реакции в этом случае в большой степени определяются давлением: при температуре выше 727 °С и давлении 0,1 МПа бимолекулярные реакции практически неосуществимы. При температуре 427 °С и таком же давлении бимолекулярные реакции протекают, но медленно— их скорость примерно в 7 раз меньше скорости распада. При температуре 427 °С и давлении I МПа бимолекулярные реакции становятся преобладающими.
Радикалы СНз, СлШ, СпНг> и СпНг,—СН2 в условиях термических процессов не распадаются. Для них существенны только бимолекулярные реакции.
12.2. ТЕРМИЧЕСКИЕ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ В ГАЗОВОЙ ФАЗЕ
Превращения алканов. Термические реакции алканов приводят к низшим алкенам и алканам. Экспериментальные данные по составу продуктов термического распада алканов хорошо объясняются радикально-цепным механизмом реакции.
Метан термически устойчив. Его термическая деструкция термодинамически возможна при температуре выше 56Q °С. Однако с заметной скоростью реакция протекает при температуре выше 1000 °С. Главными продуктами являются этан, этилен, ацетилен, углерод и водород. Первичную реакцию описывают стехиометрическим уравнением:
2СН< —> СгНб -|- Н2.
Реакция является цепной и развивается по следующей схеме (в рамках—конечные продукты):
СН3 + СН4 —>- |СШ7| + Н,
Н + СН4 —» |_НИ + СНз.
Этан менее устойчив, чем метан: его деструкция начинается при температуре приблизительно 500 °С. При 800 °С реакция идет со значительной скоростью:
СНз-СНз —>- СН2 = СН2 + Н2.
Распад по связи С—С происходит в меньшей степени.
Пропан расщепляется уже при температуре 450 °С по двум направлениям:
СН3СН = СН2 + Н2 СН2 = СН2 + СН4.
СНзСН,СН3
Начиная с бутана, распад алканов по связи С—С становится преобладающим. Относительная скорость термического крекинга алканов возрастает с увеличением молекулярной массы, что объясняется уменьшением энергии диссоциации С—С-свя-зей посредине молекулы и увеличением -числа С—С-связей с лизкой энергией диссоциации:
Число атомов уг- 5 6 7 8 10 12 20
лерода в молекуле
Относительная 1 4 9 10 32 46 120
скорость крекинга
Термический крекинг н-бутана можно представить следующей схемой. Вначале за счет разрыва связи С—С в наиболее слабом месте образуются первичные свободные радикалы (инициирование цепи):
СНзСН2СН2СНз — \~* 2СНз6Нг
СНз + СН3СН2СН
2.
Затем процесс развивается по двум возможным направлениям. По первому направлению крупные, относительно неустойчивые радикалы (Сз и выше) самопроизвольно распадаются по P-правилу с образованием более устойчивых метильных и этиль-ных радикалов или атомов водорода и соответствующих молекул алкенов:
| СНзСН = СН2 | + Н
СН3СН2СН, —
I СН2 = СН2 | + СНз.
По второму — устойчивые в отношении распада, но чрезвычайно реакционноспособные метильные и этильные радикалы и атомы водорода вступают в реакцию с исходными молекулами, отрывая от них атом водорода:
Н (СНз; С2Н5) + С4Н10 > |Н2(СН<; С2Н6) ] + С4Н9.
В результате образуются водород, метан, этан и бутильные радикалы.
Бутильные радикалы далее распадаются по р-правилу:
СН3СН2|СН2СН2 -- СН3СН2 +1сн2 — сн2
сн3|сн2снсн? —- СНэ +1сн2—СНСНз
Образующиеся при этом мелкие радикалы снова реагируют с исходными молекулами. Развивается цепной процесс. Отрыв цепи происходит путем рекомбинации и диспропорционирова-ния радикалов.
Основная часть продуктов цепной реакции образуется на стадии развития цепи, поэтому суммарный процесс крекинга н-бутана можно описать системой из двух параллельных реакций:
СН< + СН2 = СНСНз СНзСНз + СНз = СН2.
С«Н,
Термическое разложение алканов С3—С5 с увеличением глубины процесса самотормозится вследствие накопления пропилена, который реагирует с активными радикалами, ведущими цепь, с образованием малоактивного аллильного радикала:
СН3 == СН - СНз + СНз —> СНа = СН-СН3 + |СН«].
Термический распад алканов Се и выше с увеличением глубины процесса ускоряется вследствие образования алкенов, содержащих ослабленную С—С-связь в P-положении к кратной связи, что приводит к увеличению скорости инициирования.
Превращения циклоалканов. Термодинамически наиболее выгодны распад циклоалканов до элементов и дегидрирование циклопентанов до циклопентадиенов, а циклогексанов до аренов. Однако скорость этих реакций, требующих распада по С—Н-связям, на несколько порядков ниже, чем скорость крекинга по связям С—С. Поэтому главными продуктами крекинга циклоалканов являются низшие алканы и алкены, диены и водород.
Незамещенные циклоалканы распадаются по следующим направлениям:
СН2 Нзс/ \сН2
I I Н2СЧ /СН2
СН:
2
зсн2 = сн2
сн2 = сн — сн2сн2сн2сн3
СН2 = СН2 + СН2 = СНСН2СНз СНзСНз + сн2 = сн - сн = сн2.
Первичный распад молекулы происходит по одной из С—С-связей с образованием бирадикала:
/СН2 ИгС ^СН2
I -> СН2СН2СН2СН2СН2СНз
Н2С. .СН2 СН2
Бирадикал распадается на стабильные молекулы: сн2сн2сн2сн2сн2сн
|ЗСН2 = СН2|
IСН2 = СНСН2СН2СН2СНз
Реакция протекает по нецепному механизму. Скорость реакции мала ввиду высоких энергетических затрат на стадии инициирования и отсутствия низкоэнергетических-реакций продолжения цепи.
Накопление алкенов, содержащих сопряженную с двойной ослабленную С—С-связь, инициирует радикально-цепной процесс:
СН2=СНСН2-\- СН2СН2СН3 —> СН2=СНСНг + СНгСНзСНз
СН2СП2СН3 (R; Н) — >
СНзСНаСЛЫрНН; Н» | ) (1)
_!_ (2)
i 'I'"
СН2 = СН2 I + СН2 = СНСН2СН2 СН2=СНСНСН2СН2СНз
| СН2 = СН - СН = СН21 + СН2СН»
+ н-. (3>
Стабилизация радикалов СН2 = СНСНгСН2 и СН3СН2 (R) происходит путем отрыва атомов водорода от исходной молекулы (реакция (1)). При этом они превращаются соответственно в 1-бутен и этан. Кроме того, радикал С2Н5 может стабилизироваться, отщепляя атом водорода, образуя этилен. Атом водорода также атакует исходную молекулу, выделяясь в виде молекулы Нг. Образующийся по реакции (1) циклогексильный радикал превращается по реакции (2). Чередование реакций (1) и (2) представляет цепной процесс. Распад циклогексиль-ного радикала по p-связи С—Н приводит к дегидрированию (реакция 3).
Алкилциклоалканы распадаются по радикально-цепному механизму. Зарождение цепи происходит путем разрыва С—С-
связи в боковой цепи или отрыва радикала СНз от метилпро-изводных циклоалканов. Бициклические нафтены также подвергаются дециклизации, крекингу и дегидрированию.
Превращения алкенов. Алкены содержатся в исходных нефтяных фракциях и образуются при деструкции алканов и циклоалканов; их термические превращения определяют состав конечных продуктов реакции. Поэтому изучение закономерностей термических превращений алкенов представляет существенный интерес.
В условиях термических процессов при температуре 450— 500 °С термодинамически возможны реакции распада алкенов до низших алкенов, алкадиенов и алканов, образования аренов, а при более высокой температуре — ацетилена.
Низшие алкены (этилен, пропилен) в условиях термического крекинга и пиролиза подвергаются следующим превращениям.
1. Дегидрирование:
700—900° с
СНзСН = СН2 --------> СН2 = С = СН2 + 112,
-> нюо0 с
СН2 = СН2 -------- - СНишСН + На.
Механизм реакции — радикально-цепной. Инициирование реакции пиролиза этилена происходит путем разрыва а-связи С—Н, поэтому требует высокой температуры:
Н
1 < • • сн2=с^-н —> сн2=сн + н
Пропилен распадается по более слабой (3-связи С—Н:
Н
сн2=снс -j- н —*- сн2=снсн3 + н
н
Кроме того, образующийся аллильный радикал стабилизирован за счет сопряжения неспаренного электрона с л-электро-нами кратной связи. Поэтому распад пропилена происходит в значительно более мягких условиях.
Звено цепи состоит из двух реакций:
СН2 = СН —* | СН в СН | + Н,
Аналогично аллильный радикал превращается в аллен (СН2 = С = СН2).
2. Полимеризация. Реакция термодинамически возможна при атмосферном давлении до 500 °С, под давлением — и при более высокой температуре.
2СН2 = СН2 —v СН2 = СНСН2СН3.
Процесс идет через промежуточное образование бирадика-лов:
< 500° С
СН2СН2 + СН2 = СН2 —>- СН2СН2СН2СН2 —>
—> | СН2 = СНСН2СНз |.
Возможен и радикально-цепной механизм:
> 500° С
СН2 = СН + СН2 = СНз -»¦ СН2 = СНСН2СН2,
СН2 = СН2 + СН2 = СНСН2СН —» СН2 = СН +
+ I СН2 = СНСНаСНЛ.
3. Дегидроконденсация:
700° с
СН2 = СН + СН2 = СН2 ¦—> СН2 = СНСН2СН2
Н + I СН2 = СНСН=СН21
СН2=СН2+Н —>- СНг = СН + |Н2|.
Соотношение реакций дегидрирования, полимеризации и де-гидроконденсации не зависят от температуры и давления. С ростом температуры и снижением давления увеличивается выход ацетилена (реакция дегидрирования). При относительно* низкой температуре и высоком давлении идет полимеризация-низших алкенов. В промежуточных условиях — реакция дегидроконденсации.
Алкены С4—С5 кроме указанных реакций подвергаются крекингу с разрывом наиболее слабой p-связи (С—С или С—Н):
Н
-> сн2 = СН — СН = СН2 + Н2
Н2С = СН - СН - СНз р
Механизм реакций — радикально-цепной:
Н2С=СН-СН2 СНз —* СН2=СН—СН2 + СНз
СН2 = СН-СН2 —> | СН2 = С = СН2 [+н,
СН2 = СН - СНгСНз + СНз —> СН2 = СН - СНСНз + I СН4 ], СН2 = СН - СН2СНз + Й —* СН2 = СН - СНСНз + ШЛ,
СН2 = СНСНСН3 —>¦ Н 4- [ СНг= СН — СН = СН2[.
Высшие алкены при температуре 400—450 °С в реакциях дегидрирования, полимеризации и дегидроконденсации практически не участвуют, а преимущественно распадаются по р-связям С—С, образуя алкан и диен или алкены меньшей молекулярной массы:
--*• СН2=СН—CH = CH2+CH3CH8CHS
СН2 = СНСН2СН2СНаСН2СНз
> СН2 = СНСНз + СН2 = СНСН2СН3.
Первичный распад молекулы происходит по слабейшей P-связи С—С
СНг^СНСНг^СНгСНаСНгСНэ—>СН2=СНСН2 + СН2СНгСН2СН3.
311,
Далее следует стадия передачи цепи путем отрыва аллиль-ного атома водорода:
С Н2 = СНСН2СН2СН2СН2СН3 + СН2СН2СН2СН3 —>•
—-* СН2 = СН - СНСН2СН2СН2СНз' + |С«Ню|.
Звено цепи состоит из следующих реакций:
СН2=СНСНСН2->-СН2СН2СНз-> СНг=СНСН=СН2 + СНгСНгСНз,
СН2 = СНСН2СНгСН2СН2СНз + СН2СН2СН3
СН2 = СНСНСН2СН2СН2СНз + |СНзСН2СНз
Отличия в энергии Р-, у- и т. д. (кроме а-) С—Н-свя-зей значительно меньше, чем соответствующих С—С-связей, поэтому передача цепи может идти путем отрыва не только аллильного, но любого вторичного атома водорода. Вероятность отрыва других водородных атомов возрастает с увеличением длины цепи алкена (статистический фактор):
СН2 = СНСН2СН2СН2СН2СН3 + R3 —->¦
—->¦ rh + ch2 = chch2ch2ch2chch3>
CH2=CHCHzCH2| СНгСНСНз —СН2 = СНСНгСНг + jCH2—СНСщ], СН2 = СНСН2СН2СН2СН2СНз + сн2 = СНСН2СН2 —
—>- СН2 = СНСН2СН2СН2СНСНз + СН2 = СНСН2СНз |
Состав конечных продуктов крекинга алкена зависит также от возможной изомеризации радикалов.
Таким образом, цепной процесс крекинга высших алкенов в определенной мере протекает независимо от наличия двойной связи.
Циклоалкены более устойчивы, чеь? алкены. Циклогексен устойчив до 600°С, при более высокой температуре он распадается по нецепному механизму через бирадикал или цепным путем дегидрируется в бензол.
Превращения алкадиенов и алкинов. При относительно невысокой температуре (ниже 400 °С) и давлении, близком к атмосферному и выше, алкадиены более реакционноспособны, чем соединения других классов. Основное направление реакции— диеновый синтез, протекающий по молекулярному меха-
низму:
+
I!
С
/СН=СН2
сн2
СН = сн2
/
СН
II
сн2
нс^ Ht
+
\:н2
При температуре выше 700 °С и атмосферном давлении диеновый синтез протекает в незначительной степени, основное значение приобретает радикально-цепное превращение (крекинг, дегидроконденсация и др.).
Цепной распад ацетилена можно представить схемой:
2СН = СН —-> СН = С + СН = СН2 CHsC + CHesCH—> сн = ссн = сн
СНэСН
СН зз ссн = СН —
сн^сн
|СН = С — С ¦= СН| + Й |CHs=CCH = CH2 + СН 55 С ¦ СН = ССН = СНСН - СН
CHsCH
сн^с
В результате термического превращения алкадиенов и ацетилена образуются сильноненасыщенные соединения большей молекулярной массы и арены.
При конденсации аренов и алкенов (алкинов) получают замещенные и поликонденсированные арены:
fol + 2CHs=CH —> СН = СН — СН = СН2 —v
Дегидрокондснсация алкенов и алкадиенов, диеновый синтез и циклизация силыюненасыщенных соединений являются промежуточными ступенями, ведущими к ароматизации продук-
тов термических превращений углеводородов нефтяных фракций.
Превращения аренов. Термическая устойчивость аренов сильно изменяется в зависимости от строения. Незамещенные и метилзамещенные бензол и нафталины значительно более устойчивы, чем алканы. Термодинамически возможен распад незамещенных аренов до элементов, а при очень высокой температуре— раскрытие аренового кольца. Однако, исключая электрокрекинг, который протекает при очень высокой температуре, незамещенные арены подвергаются практически только дегидроконденсации. Алкилзамещенные арены, имеющие связь С—С, сопряженную с кольцом, разлагаются быстрее алканов. Это объясняется распределением энергии между связями в молекуле (см. раздел 12.1). Основным направлением превращения алкил-ареновых углеводородов является крекинг алкановых цепей и деалкилирование.
В условиях термических процессов осуществляется дегидро-конденсация бензола с образованием дифенила и водорода:
2СбНб —> СбНб — СеНб -|- Нг
Механизм реакции — радикально-цепной:
СеНе •—> CeHs -|- Н
Н -f- СвНб —>• | Нг | -)- у-Аналогично происходит дегидроконденсация нафталина:
Реакции дегидроконденсации аренов вместе с диеновым синтезом и дегидроконденсацией алкенов лежат в основе процессов коксообразования при термическом крекинге и пиролизе. Толуол, как и бензол, подвергается дсгидроконденсации:
CeHjCHsCHsCeHs
СвН5СН2СвН4СНз.
За счет водорода, выделившегося при дегидроконденсацииг частично протекает деметилирование:
СвН5СНз -|- Нг •—> СеНе -|- СН4.
В начальный период превращение толуола происходит с небольшой скоростью, так как радикальная реакция идет по нецепному пути:
СбНвСНз —>- CeHsCH2 + н —> ГнЛ + сбн5сн2
(о) +СН3
![]()
![]()
СНз + <Ъ^)—СНз —V | сн41 + с6н5сн2 2С6Н5СН2 —>- С„Н5СН2 - СН2С„Н6
Бензильный радикал (СбНбСНг) малоактивен и вступаег главным образом в реакции рекомбинации с образованием дибензила, вследствие чего цепь обрывается.
С увеличением глубины процесса повышается концентрация дибензила. За счет распада алифатической С—С-связи в дибензиле и развития цепного механизма скорость превращения: толуола возрастает:
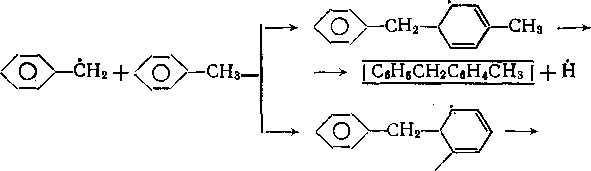
СНз
—* I С8Н5СН2СбНГ| + СНз СНз (Й) + СвН5СН3 —>- | СН4 (Н2) | + С„Н6СН2 2СвН6СН2 | СбН6СНаСН2СбН61
Алкилпроизводные аренов с длинными боковыми цепями в условиях термических процессов подвергаются распаду алкильных цепей:
Инициирование реакции происходит путем разрыва слабейшей p-связи С—С, сопряженной с ароматическим кольцом:
C6H5—СНг^СНг—СН2СН3 —> С6Н5СН2 + СН2СН2СН3
При реакции малоактивного бензильного радикала с исход-дым углеводородом происходит отрыв наиболее слабо связан-лого атома водорода от а-углеродного атома алкановой цепи:
С8Н5СН2СН2СН2СН3 -|- свн5сн2 —>¦
-—> С6Н5СНСН2СН2СН3 -|- j С6Н5СН31.
При взаимодействии активного алкильного радикала (С3Н7) возможен отрыв любого атома водорода боковой цепи.
Распад вновь образовавшегося радикала приводит к появлению арена с алкеновым заместителем и низкомолекулярного алкильного радикала:
СбН5СНСН2СН2СН3 —> | С6Н5СН = СН2 | + СН2СНз.
Реакция радикала C2Hs с исходным углеводородом превращает его в конечный продукт СгНб и замыкает цепь. При распаде радикалов по P-связи С—Н выделяется водород. Присоединение атома водорода к ареновому кольцу ведет к деалки-лированию алкиларена:
H + <o>-R
+ R-
\2/
ОС
Таким образом, основными продуктами термического превращения алкиларенов являются стирол, алкан, другие продукты крекинга алкановой цепи, а также в меньшей степени толуол и бензол.
Превращения смеси углеводородов. Цепные реакции, протекающие в системе, всегда взаимосвязаны. Скорость инициирования радикально-цепного превращения смеси определяют реакции распада на радикалы наименее стабильных компонентов, таких как алкены или алкиларены, содержащие ослабленные P-связи С—С. Инициирование является самой энергоемкой стадией цепного процесса. Дальнейшие превращения радикалов происходят обычно с очень небольшими энергиями активации. Вследствие увеличения скорости инициирования термический распад смеси углеводородов во многих случаях протекает с большей скоростью, чем распад индивидуальных соединений. Отдельные углеводороды, распадающиеся в чистом виде по нецепному пути из-за малой скорости инициирования, в смеси могут превращаться по цепному механизму. Таким образом, термические реакции смеси углеводородов осуществляются преимущественно цепным путем.
Крекинг углеводородов, таких как бензол, толуол, этилен, пропилен, образующих радикалы, стабильные к распаду, в смеси с другими углеводородами (алканами, циклоалканами) будет сильно тормозиться. Например, фенильный радикал при разложении бензола присоединяется к молекуле бензола, образуя дифенильный радикал:
СбН6 -)- СбН6 —v СбН6СбНб-
Если же бензол находится в смеси с алканами, то для фе-нильного радикала возможна и такая реакция:
CeHs + RH —> СбНб + R-
Так как скорость взаимодействия фенильного радикала с алканом выше, чем с бензолом, последний практически расходоваться не будет.
В результате взаимного влияния углеводородов на стадии развития цепи состав продуктов термического распада смеси углеводородов отличается от продуктов распада индивидуальных веществ.
12.3. ПИРОЛИЗ
Закономерности термического разложения углеводородов в определенной мере изменяются при переходе от условий термического крекинга (470—540 °С) к условиям пиролиза (700— 1000 °С). Температура влияет на механизм процесса и на состав продуктов.
Суммарные реакции, протекающие при пиролизе и крекинге, можно разделить на три основные группы: 1) первичные реакции крекинга и дегидрирования, приводящие к образованию алкенов; 2) вторичные реакции превращения алкенов —полимеризация и конденсация; 3) реакции прямого молекулярного распада, при котором образуются пироуглерод, водород и частично ацетилен.
В условиях высоких температур пиролиза при очень значительной энергонасыщенности молекул возрастает концентрация радикалов. Это приводит к уменьшению длины цепи и увеличению роли радикально-нецепного разложения, при котором отдельные углеводороды разлагаются независимо друг от друга.
Повышение температуры ускоряет реакции с более высокими значениями энергии активации, вследствие чего меняется соотношение между различными реакциями радикалов. Возрастает значение более энергоемких реакций распада радикалов по сравнению с менее энергоемкими реакциями присоединения. Температура влияет также на вторичные реакции превращения алкенов. Распад алкенов, протекающий с высокими энергиями активации, значительно ускоряется с повышением температуры по сравнению с реакциями конденсации алкенов, характеризующимися более низкими значениями энергии активации. И, наконец, температура определяет соотношение между основными группами реакций пиролиза (первичными, вторичными и образованием пирюуглерода). Значения энергий активации этих типов реакций можно расположить в ряд:
?3 > Ei > ?2,
где Е\—энергия активации первичных реакций; Е2—'Энергия активации вто-рячных реакций; Е3—энергия активации элементного распада.
Если целевым назначением термического процесса является получение алкенов, то реакцию необходимо проводить при высокой температуре, с тем чтобы скорость первичных реакций была выше скорости вторичных процессов. Однако поднимать температуру выше 900 °С нецелесообразно, так как при этом с заметной скоростью начинают протекать реакции распада.
Для получения низкомолекулярных алкенов процесс необходимо проводить при пониженном давлении. Однако технологические особенности процесса, требующие больших скоростей сырьевого потока для обеспечения малой продолжительности реакций, связаны с преодолением значительных гидравлических сопротивлений, для чего создают повышенное давление на входе в реакционный змеевик. Снижения давления углеводородов добиваются разбавлением сырья инертными веществами (обычно водяным паром).
Скорость пиролиза углеводородов увеличивается в присутствии молекулярного водорода. Метальный радикал, ведущий цепной процесс пиролиза наряду с атомарным водородом, в присутствии молекулярного водорода реагирует по двум параллельным реакциям — с молекулой водорода и исходным углеводородом, например гексаном:
СН3 + Н2 —> СН< + Н,
СНа + СвН,4 —v СН4 + СвН13.
При температуре 827°С константа скорости первой реакции на порядок выше второй (при равных концентрациях На Щ С6Нн). Скорость реакции метального радикала с алкенамйЦ также ниже, чем скорость взаимодействия с водородом (для
1-бутена константа скорости отличается в 4 раза). ==
Образующийся атомарный водород реагирует с углеводо*| родными молекулами сырья. Константа скорости этой реакцн*а на 2—3 порядка больше, чем константа скорости взаимодейст-J вия углеводородов с метальным радикалом. В результате молекулярный водород играет роль гомогенного катализатора суммарного процесса пиролиза. Кроме того, он подавляет в значительной степени реакции образования диенов, реагируя с ви-нильными радикалами (СНг = СН) и предотвращая их присоединение к этилену. Следствием этого является снижение выхода тяжелых продуктов конденсации.
12.4. ОСОБЕННОСТИ ТЕРМИЧЕСКИХ РЕАКЦИИ
В ЖИДКОЙ ФАЗЕ
При атмосферном давлении в 1 см3 газа содержится приблизительно 1019, а в таком же объеме жидкости — примерно 1021 молекул. Концентрация молекул в жидкости такая, как в газе под давлением 10 МПа. Поэтому проведение реакций в жидкой фазе с точки зрения соотношения скоростей моно- и бимолекулярных реакций равносильно проведению их в газовой фазе под высоким давлением. В результате при равных температурах жидкофазные термические реакции углеводородов и нефтепродуктов приводят к значительно большему выходу продуктов конденсации и меньшему выходу продуктов распада. На суммарный результат превращения углеводородов в жидкой фазе определенное влияние оказывают «клеточный эффект» и сольватация. При распаде молекулы углеводорода на радикалы в газовой фазе последние немедленно разлетаются. В жидкой фазе радикалы окружены «клеткой» из соседних молекул. Для удаления радикалов на расстояние, при котором они становятся кинетически независимыми частицами, необходимо преодолеть дополнительный активационный барьер, равный энергии активации диффузии радикала из клетки. С другой стороны, и для рекомбинации радикалы должны преодолеть клеточный эффект. В результате суммарная концентрация радикалов в жидкости останется такой же, как и в газовой фазе. Однако, если радикалы существенно различаются по массе и активности, то клеточный эффект может изменить стационарную концентрацию радикалов, что приведет к изменению энергии активации жидкофазной реакции относительно газофазной.
Сольватация существенно влияет на скорость взаимодействия полярных частиц. Скорость жидкофазных радикальных реакций практически от сольватации не зависит.
12.5. ОБРАЗОВАНИЕ НЕФТЯНОГО КОКСА
Кокс образуется в жидкофазных термических процессах. В ряде случаев, например при термическом крекинге, он является нежелательным побочным продуктом. С другой стороны,
существуют процессы, разработанные специально для получения кокса, имеющего важное народнохозяйственное значение. Его применяют в качестве восстановителя в химической технологии, для приготовления анодов в металлургии, в авиационной и ракетной технике (ВегС; TiC), в производстве абразивов и огнеупоров (SiC; В4С; TiC), в ядерной энергетике (В4С, ZrC), для получения конструкционных углеграфитовых материалов.
Нефтяной кокс — это твердое вещество плотностью 1400— 1500 кг/м3 с высоким содержанием углерода. Атомное отношение С : Н в коксе составляет 1,1—4. Он образуется при жидкофазной термической переработке нефтяных остатков по схеме:
Арены —v Смолы -—>¦ Асфальтены —»¦ Кокс -—> Графит.
Алканы, циклоалканы и алкены также способны к коксооб-разованию, но не непосредственно, а в результате глубоких превращений и ароматизации.
Переход аренов в кокс и далее в графит термодинамически закономерен, так как он сопровождается снижением уровня свободной энергии. В ряду бензолнафталин-*- антрацен ->- пирен ->- графит запас свободной энергии (в кДж) на один атом углерода уменьшается в следующем порядке: 20,6—>- 19,8-»-—> 18,8—> 16,8—*-0.
Сырьем для получения кокса служат тяжелые нефтяные остатки. Их состав и строение молекул достаточно подробно рассмотрены в гл. 11.
При коксовании (400—500 °С) протекают следующие превращения основных компонентов тяжелых нефтяных остатков (масел, смол, асфальтенов).
Парафино-нафтеновая часть масел крекируется до жидких и газообразных продуктов. Моно- и бициклоароматические углеводороды масел реагируют двумя путями. Как и парафино-наф-теновые углеводороды, они могут подвергаться крекингу. Вместе с тем, благодаря наличию в молекулах нафтеновых циклов с подвижными атомами водорода, становятся возможными реакции перераспределения водорода. В результате этого процесса часть молекул превращается в насыщенные углеводороды и крекируется, другая часть становится более ароматичной и пополняет твердую фазу асфальтенов.
Смолы частично крекируются до газообразных и жидки* продуктов. Основная же часть смолистых компонентов деалкщ лирустся и теряет кислородсодержащие функциональные груп-~ пы. Вследствие этого повышается степень ароматичности, и смолы превращаются в асфальтены.
Асфальтены при температуре выше 300°С разлагаются с об* разованием газа, жидких продуктов и кокса. В нефти и et;j остатках асфальтены благодаря наличию сольватных оболочек
находятся в тонкодисперсном состоянии. В условиях коксования асфальтены лишаются сольватной оболочки (которая подвергается крекингу), теряют алкильные заместители и функциональные группы. Расстояния между молекулами сокращаются, ассоциаты становятся более компактными, увеличивается энергия их дисперсного притяжения, вследствие чего они последовательно по мере потери водорода переходят в карбены, карбоиды и кокс.
По данным рентгенографического анализа, кокс представляет собой агломерат хаотично расположенных трехмерно неупорядоченных кристаллитов, сходных по строению и размерам с ассоциатами асфальтенов, но с меньшим межслоевым расстоянием (0,348—0,350 н. м.). Начиная с 400 °С увеличивается толщина слоистоблочных структур, что объясняют достройкой кристаллитов за счет вновь образующихся молекул асфальтенов.
Наличие в коксе водорода, а возможно, и метильных заместителей не дает возможности образоваться трехмерно упорядоченной структуре, подобной графиту. Такое упорядочение наступает при температуре выше 1200—1500 °С, когда удаляется почти весь водород.
12.6. ПРОМЫШЛЕННЫЕ ПРОЦЕССЫ
ТЕРМИЧЕСКОЙ ПЕРЕРАБОТКИ НЕФТИ И НЕФТЯНЫХ ФРАКЦИЙ
Основные процессы термической переработки нефти — термический крекинг, пиролиз и коксование.
В зависимости от условий проведения термического процесса сырье находится в различных агрегатных состояниях: пиролиз протекает как газофазная реакция, коксование нефтяных остатков происходит в жидкой фазе, при термическом крекинге тяжелого сырья возможно сосуществование газовой и жидкой фаз.
Термический крекинг. Процесс термического крекинга в промышленности применяют с 1912 г. Его первоначальным назначением было получение автомобильного бензина. Однако из-за возросших требований к качеству моторного топлива к 60-м го-дам он был полностью вытеснен каталитическим крекингом.
В настоящее время термический крекинг тяжелых остатков переработки нефти проводят с целью получения вакуумного газойля (термогазойля) или маловязкого котельного топлива (мазута — крекинг-остатка).
Существуют разнообразные схемы термического крекинга. Вариант термического крекинга, направленный преимущественно на снижение вязкости котельного топлива, получил в мировой практике название «висбрекинг» (легкий крекинг). Процесс осуществляют при температуре 450—480 °С под давлением 2—5 МПа. При этом происходит частичное удаление нестабильных серосодержащих соединений (сероводорода, сульфидов, дисульфидов). Сырьем служат нефтяные остатки — полугудроны, гудроны, асфальты, экстракты, тяжелые газойли каталитического крекинга. Основные продукты висбрекинга — углеводе^ родный газ, крекинг-бензин, керосино-газойлевая фракция, термогазойль и крекинг-остаток.
Ниже приведен выход продуктов (в %) в процессе термического крекинга357 с максимальным выходом крекинг-остатка(I) и термогазойля(II):
Г аз термического крекинга, представленный главным образом углеводородами Ci—Сз, содержит значительное количество непредельных углеводородов (см. табл. 10.1). Его применяют в качестве нефтехимического сырья или как топливо.
Бензин также содержит много алкенов, вследствие чего он характеризуется низкой химической стабильностью. Наличие сернистых и азотистых соединений, невысокое октановое число (60—66 по моторному методу) не позволяют использовать крекинг-бензин в качестве компонента моторного топлива без предварительной гидроочистки и риформирования.
Керосино-газойлевая фракция (200—350 °С) является ценным компонентом флотского мазута. После гидроочистки ее применяют также как компонент дизельного топлива.
Термогазойль (>350 °С)—сырье для каталитического крекинга (гидрокрекинга) и производства технического углерода. Крекинг-остаток используют как котельное топливо. По сравнению с прямогоиным мазутом продукт вследствие арома* тизации имеет ббльшую плотность и теплоту сгорания. Для получения маловязкого котельного топлива в крекинг-остатке оставляют небольшое количество сравнительно иизкомолекуляр^ ных газойлевых фракций. При соответствующем изменении технологии висбрекинга можно понизить также температуру застывания остатка. Ниже приведена характеристика остатка
Исходное Остаток
сырье висбрекинга
Висбрекинг является в настоящее время одним из перспективных процессов глубокой переработки высоковязких нефтяных остатков. Включение его в схему переработки нефти позволяет значительно увеличить отбор вакуумного газойля и получать товарное котельное топливо без применения разбавителей.
Пиролиз. Основное назначение процесса пиролиза углеводородного сырья — получение низших алкенов. Процесс проводят при 800—900 °С под давлением, близким к атмосферному. Для снижения парциального давления углеводородов сырье обычно разбавляют водяным паром. Оптимальным сырьем для производства этилена является этан. Выход этилена при этом достигает 80%. Значительный выход этилена наблюдается также при пиролизе алканов нормального строения: из пропана — до 48 %, из бутана — 45 %. При пиролизе разветвленных алканов образуются преимущественно алкены Сз—С4 и алкадиены, а при высокой температуре — также аллен и метилацетилен. Выход низших алкенов при пиролизе циклоалканов и аренов невелик.
Важнейшим фактором при выборе сырья пиролиза является доступность, что в разных странах определяется сложившимися способами переработки нефти и газа. В США до 70 % общего объема этилена вырабатывают из газообразных углеводородов, преимущественно из этана, природного и попутного газов. В СНГ, странах Западной Европы и Японии, напротив, основную часть этилена получают пиролизом прямогонных бензинов и газойлей. При пиролизе бензинов наряду с алкенами С2—С4 и бутадиеном образуется метановодородная фракция, значительное количество жидких продуктов, содержащих алкены, циклоалкены, алкадиены, арены и другие компоненты. Выход продуктов при пиролизе бензинов различного состава колеблется в широких пределах, %:
При пиролизе керосино-газойлевых фракций выход этилена составляет 16—23 %. пропилена — около 15 %, жидких продуктов — примерно 50 %.
В связи с непрерывным ростом цен на прямогонные бензины и их недостаточными ресурсами в балансе сырья пиролиза ожидается увеличение доли природного и попутного газов, а также бензиновых фракций, выделенных из газовых конденсатов. Все более широкое применение как сырье пиролиза находят вторичные продукты нефтехимии. Главным образом это относится к бензинам-рафинатам, получаемым после выделения из бензинов риформинга ароматических углеводородов. Газоконденсатный бензин по сравнению с прямогонным содержит повышенное количество аренов, а бензин-рафинат — изоалканов, поэтому выход этилена из них примерно на .10 % ниже, чем из прямогонного бензина.
В настоящее время наблюдается устойчивая тенденция вовлечения в процесс пиролиза также более тяжелого углеводородного сырья. Это сырье содержит много конденсированных аренов и циклоалканов (до 40—50 %), что приводит к повышенному закоксовыванию змеевика, снижению выхода этилена и большому количеству тяжелых фракций. Пиролиз такого сырья осуществляют в сравнительно мягких условиях: температура 800—820 °С, время контакта 0,4—0,5 с — и при большом разбавлении сырья водяным паром (до 80—100 %). Состав образующихся продуктов представлен в табл. 12.1.
Для интенсификации процессов пиролиза тяжелых нефтяных дистиллятов производят их предварительную гидрокаталитическую обработку: гидроочистку, гидродеароматизацию, гидрокрекинг и экстрактивную деароматизацию. Уменьшение содержания полициклических аренов снижает коксообразование и позволяет вести процесс в более жестких условиях. При пиро-
Таблица 12.1. Состав продуктов пиролиза углеводородного сырья
| Бензин | Вакуумный газойль | ||
| Компонент | прямой перегонки | исходный |
после гидрокрекинга |
|
сн4 | 15,6 |
7,6 | 10,1 |
|
с2н4 | 26,0 |
17,6 | 26,2 |
|
СзН6 | 14,0 |
10,9 | 13,1 |
|
QHg | 4,3 | 4,0 |
4,4 |
|
с4н6 | 3,8 |
3,5 | 4,7 |
|
Пирокоидеисат | 23,1 |
17,0 | 19,8 |
|
в том числе | |||
| беизол | 7,0 | 3,0 |
5,0 |
| толуол |
4,5 | 2,0 | 2,2 |
| Тяжелая смола (> 200°С)* |
5,8 | 34,1 | 16,6 |
| * Тяжелая смола пиролиза содержит нафталин, метилнафталииы, дифенил, флуорен, фенаитреи, антрацен, ацеиафтеи. |
|||
лизе деароматизированного сырья получают почти столько же алкенов, сколько из прямогонного бензина (табл. 12.1). При сравнении различных схем подготовки вакуумного газойля как сырья для пиролиза отечественными исследователями показано, что предпочтительной является схема глубокого гидрирования на первой ступени гидрокрекидга под давлением водорода около 15 МПа.
Таким образом, наилучшим сырьем для получения этилена и пропилена являются газообразные углеводороды Сг—С4. Однако ассортимент других продуктов несопоставимо беднее, чем при пиролизе бензина и более высококипящих фракций. Так как стоимость сырья составляет около 70 % себестоимости этилена, выбор сырья является важной экономической задачей и определяется в целом его доступностью, стоимостью и возможностью реализации всех сопутствующих продуктов.
Необходимость расширения сырьевой базы, сокращения расхода сырья, а также удельных энергетических и материальных затрат привела к разработке новых модификаций процесса, рассчитанных в основном на пиролиз тяжелых видов углеводородного сырья. К числу принципиально новых процессов относят в первую очередь следующие: пиролиз в присутствии гетерогенных катализаторов (каталитический пиролиз); пиролиз в присутствии гомогенных инициирующих добавок; высокотемпературный пиролиз с использованием газообразных теплоносителей; пиролиз в расплаве металлов и их солей; термоконтактные процессы.
Наибольшую активность из исследованных катализаторов в процессе каталитического пиролиза проявляют ванадат калия, оксид индия, оксидный железохромовый (88 % РегОз+ + 7 % СггОз) и др. Для снижения коксообразования в состав катализатора рекомендуют вводить модификаторы: К2СО3,
K2SO4, Fe(N03)2 и Н3ВО3. Исходное сырье, как и в обычном пиролизе, разбавляют водяным паром, однако в каталитическом процессе водяной пар не только снижает парциальное давление углеводородов сырья, но и участвует в реакциях разложения углеводородов, увеличивая степень превращения. Предполагают, что вода подвергается диссоциативной адсорбции на поверхности катализатора и дополнительно генерирует активные радикалы. Гетерогенно-каталитический пиролиз прямогонных бензинов протекает при более низкой температуре, чем некаталитический (780 °С вместо 830—840 °С), и дает суммарный выход алкенов почти на 10 % выше, чем обычный пиролиз (60—63 % вместо 53%). Каталитический пиролиз более тяжелых нефтяных фракций, таких как вакуумный газойль, рекомендуют проводить с предварительным гидрокрекингом.
В инициированном процессе в качестве гомогенных инициаторов реакций пиролиза изучен и предложен ряд веществ: галогены и галогенсодержащие вещества (главным образом, НС1), пероксиды водорода и органических веществ, сера и серосодержащие вещества, водород и соединения, образующие водород при термическом разложении. Применение инициаторов позволяет ускорить первичные реакции разложения сырья и увеличить выход этилена. Пиролиз в присутствии водорода (гидропиролиз) рекомендуют проводить под давлением водорода 2,0—2,5 МПа. Во избежание гидрокрекинга алкенов температура должна быть 800—900 °С при малом времени контакта— около 0,1 с. Водород действует как инициатор процесса, увеличивает выход этилена, а также снижает коксообразование и выход тяжелых фракций пироконденсата. Недостаток этого процесса — значительный расход водорода и увеличение выхода метана. Гидропиролиз прямогонного бензина позволяет получать до 40—45 % этилена. Выход метана при этом достигает 34 %, пиробензина — 20 %, тяжелой фракции пироконденсата —
2—3 %. В качестве сырья можно использовать тяжелые нефтяные фракции (вакуумный газойль и др.), а также фракции со значительным содержанием алкенов и даже аренов.
К перспективным методам переработки тяжелых видов сырья относят также процессы пиролиза с использованием теплоносителей: газообразных (водяной пар, дымовой газ, водород), жидких (расплавы металлов Pb, Bi, Cd, Sn и др., а также их сплавы и соли) и твердых контактов (мелкозернистый кокс, песок). Эти процессы находятся в стадии исследования и опытно-промышленных испытаний.
Коксование. Назначение процесса коксования — получение нефтяного кокса и дистиллята широкого фракционного состава.
В качестве сырья для нефтяного кокса могут быть использованы отбензиненные нефти; остатки первичной переработки — мазуты, полугудроны; продукты вторичного происхождения — крекинг-остатки, тяжелые газойли каталитического крекинга, смолы пиролиза, а также природные асфальты и отходы масляного производства (асфальты, экстракты).
Существует несколько - модификаций процесса: периодическое коксование в кубах, замедленное коксование в необогре-ваемых камерах, коксование в псевдоожиженном слое порошкообразного кокса. Наибольшее распространение получил полунепрерывный процесс в установках замедленного коксования.
Замедленное коксование нефтяных остатков протекает при температуре 490—505 °С и давлении 0,2—0,3 МПа. В результате коксования кроме нефтяного кокса получают газ, бензин, средние и тяжелые коксовые дистилляты. Выход продуктов и их качество зависят от химического и фракционного состава сырья и условий коксования.
Выход кокса из остатка первичной переработки нефти составляет 15—25%, из вторичных продуктов — 30—35 %. Вместе с коксом образуется значительное количество денных жидких и газообразных продуктов, свойства которых близки к характеристикам продуктов термического крекинга. Их суммарный выход достигает 70 % (масс.) в расчете н,а исходное сырье.
Коксование тяжелых нефтяных остатков является одним из наиболее экономичных способов превращения их в дистиллят-ное сырье. Наибольшая эффективность процесса коксования наблюдается при квалифицированном Использовании всех образующихся продуктов.