Глава 6 исследование состава нефти и нефтепродуктов
Глава 6
ИССЛЕДОВАНИЕ СОСТАВА НЕФТИ И НЕФТЕПРОДУКТОВ
Химический и фракционный состав нефтей необходимо знать для выбора наиболее рационального комплекса процессов нефтепереработки, их моделирования, обоснования мощности нефтеперерабатывающих установок, а также для развития представлений о генезисе нефти и решения задач нефтяной геологии.
Различают несколько видов анализа нефтей и нефтяных фракций: элементный, индивидуальный, групповой, структурногрупповой. Развитие техники современных физико-химических методов анализа смесей позволило перейти от определения элементного состава нефтей к исследованиям группового и индивидуального состава нефтяных фракций. Разработаны методы изучения индивидуального состава газа и бензиновых фракций (до Сю), группового состава и идентификации ряда индивидуальных компонентов керосино-газойлевых фракций (до Сго).
При анализе масляных фракций и смолисто-асфальтеновых составляющих нефтей удается идентифицировать пока лишь некоторые индивидуальные соединения. Групповое разделение этих фракций, включающих гибридные структуры, — также достаточно сложная и не вполне решенная задача. С использованием масс-спектроскопии, ЯМР-спектроскопии и других современных методов проводят структурно-групповой анализ высокомолекулярных нефтяных фракций: определяют содержание
углерода в алифатических, алициклических и ароматических структурах, содержание водорода в водородсодержащих фрагментах, среднее число ароматических и насыщенных колец и т. д.
В России приняты единые унифицированные программы исследования нефтей: основная — для нефтей новых месторождений или новых горизонтов действующих месторождений, имеющих большое промышленное значение или уникальных по составу, а также две сокращенные программы — для исследования нефтей новых малодебитных месторождений и нефтей из разведочных скважин.
Для правильного выбора метода переработки нефти, составления материальных балансов некоторых процессов необходимо знать элементный состав нефти.
Наличие в нефти серо- и кислородсодержащих соединений требует сооружения специальных установок очистки. Для этого необходимы сведения о содержании в нефти серы и кислорода. Серосодержащие соединения наиболее вредны как при переработке нефти, так и при использовании нефтепродуктов; поэтому содержание серы входит как показатель в ГОСТ на нефть.
Массовое содержание серы, кислорода и азота в нефти, казалось бы, невелико и в сумме редко превышает 3—4%. Однако на каждую единицу массы этих элементов приходится 15—20 единиц массы углеводородных радикалов, откуда на долю углеводородной части нефти приходится только 40—50 % от общей массы нефти.
Основную часть нефти и нефтепродуктов составляют углерод (83—87 %) и водород (12—14%). Их содержание, а иногда и соотношение полезно знать для расчетов некоторых процессов. Например, процентное отношение массового содержания водорода к содержанию углерода (100 Н/С) показывает, сколько необходимо добавить водорода к сырью в процессе гидрогенизации (гидрокрекинга), чтобы получить желаемые продукты. Отношение 100 Н/С в бензине равио 17—18, в нефти 13—15, в тяжелых фракциях 9—12.
При каталитическом крекинге происходит диспропорциони-рование водорода между продуктами реакции. В идеальном процессе крекинга (когда весь водород сырья переходит в бензин) из нефти можно получить 75—80 % бензина. На самом деле в промышленных условиях за счет газообразования и реакций уплотнения выход бензина снижается до 40—50 %.
Данные об элементном составе нефти и нефтепродуктов необходимы для расчета таких процессов, как горение, газификация, гидрогенизация, коксование и др.
Данные элементного и структурно-группового состава узких фракций масел и тяжелых остатков, из которых выделение индивидуальных соединений практически невозможно, позволяет значительно расширить представления о структуре веществ, входящих в эти фракции, и построить модель их «средней» молекулы.
Более подробные сведения о содержании элементов, особенно серы, азота и кислорода, в нефтях и нефтепродуктах приводятся в гл. 11.
Элементный анализ на углерод и водород основан на без остаточном сжигании органической массы нефтепродукта в токе кислорода до диоксида углерода и воды. Послед-
ние улавливают и по их количеству рассчитывают содержание указанных элементов. Необходимо, чтобы горение было полным (образующийся СО окисляют до С02), а продукты сгорания были очищены от оксидов серы, галогенов и других примесей.
Определение серы можно проводить различными методами. Для легких нефтепродуктов применяют ламповый метод или сжигание в кварцевой трубке. Для средних и тяжелых нефтепродуктов пригоден метод смыва конденсата при сжигании образца в калориметрической бомбе.
Сущность лампового метода заключается в сжигании нефтепродукта некоптящим пламенем в специальной лампе и' улавливании образовавшегося диоксида серы в абсорберах с раствором соды. Последующим титрованием избытка соды определяют ее количество, пошедшее на связывание диоксида серы, и вычисляют количество серы (ГОСТ 19121—73).
Метод сжигания в трубке принципиально ничем не отличается от лампового метода, только образовавшийся в процессе горения диоксид серы окисляют пероксидом водорода до триоксида серы; дальнейшее определение ведут как в предыдущем методе.
Принцип метода смыва бомбы заключается в сжигании нефтепродукта в калориметрической бомбе, в которую предварительно залито 10 см3 дистиллированной воды. После сжигания воду из бомбы и смывы ее со стенок и других деталей переносят в колбу, подкисляют, кипятят для удаления СОг, затем добавляют хлорид бария. Выпавший осадок сульфата бария выделяют, сушат и по его массе вычисляют содержание серы.
Содержание азота определяют методом Дюма или Кьель-даля. Метод Дюма основан на окислении нефтепродукта твердым окислителем — оксид меди(П) — в токе диоксида углерода. Образовавшиеся в процессе окисления оксиды азота восстанавливают медью до азота, который улавливают после поглощения С02, и по его объему определяют количество азота в нефтепродукте. По методу Кьельдаля нефтепродукт окисляют концентрированной серной кислотой. Из образующегося сульфата аммония азот выделяют при обработке щелочью в виде аммиака, который улавливают титрованным раствором кислоты.
Процентное содержание кислорода чаще всего определяют по разности между 100 и суммарным содержанием всех остальных элементов в процентах. Это неточный метод, так как на его результатах сказываются погрешности определения всех остальных элементов. Имеются прямые методы определения кислорода, например гравиметрический метод пиролиза нефтепродуктов и токе инертного газа в присутствии платинированного графита и оксида меди. О содержании кислорода судят по массе выделившегося СОг.
6.2. ОПРЕДЕЛЕНИЕ ГРУППОВОГО СОСТАВА
Даже узкие фракции нефти представляют собой сложные смеси углеводородов и гетероатомных соединений.
Узкие бензиновые и даже керосиновые фракции можно разделить на индивидуальные углеводороды с помощью газожидкостной хроматографии. Несмотря на относительную быстроту хроматографического анализа, расшифровка и расчет хроматограмм таких сложных смесей очень трудоемки. Для технических целей часто нет необходимости в таком детальном анализе. Достаточно знать суммарное содержание углеводородов по классам.
Уже сравнительно давно в практике нефтепереработки существуют методы определения состава нефтепродуктов по содержанию в них тех или иных классов углеводородов (групповой состав для бензинов и структурно-групповой состав для масел и тяжелых остатков нефти). Эти методы можно подразделить на следующие типы: химические, физикохимические, комбинированные и физические.
Химические методы предусматривают взаимодействие реагента с углеводородами определенного класса (аренами или алкенами), о наличии которых судят по изменению объема или количеству образовавшихся продуктов реакции. К ним относятся, например., нитрование и сульфирование.
Физико-химические методы включают экстракцию и адсорбцию, например экстракцию аренов диоксидом серы, диметилсульфатом, анилином и т. п. и адсорбцию этих углеводородов на силикагеле.
Комбинированные методы наиболее точны и широко распространены. Они основаны на совместном использовании каких-либо двух методов: удаляют арены химическим или физико-химическим методом и измеряют физические свойства нефтепродукта (плотность, показатель преломления, изменение критических температур растворения в других жидкостях и др.) до и после удаления аренов.
Физические методы основаны главным образом на определении оптических свойств.
Анализ группового состава масляных фракций несколько сложнее. С повышением молекулярной массы нефтепродуктов в них все большую долю составляют гибридные структуры и различия между классами углеводородов стираются. В этом случае задачей анализа является не только определение количества аренов, циклоалканов и алканов в продукте, но и изучение гибридных соединений по содержанию в них различных структурных единиц (ароматических и алициклических колец, алкильных заместителей).
Приемы для таких анализов используются те же — комбинированное применение физико-химических, химических и физических методов исследования, а также использование эмпирических уравнений и номограмм.'
Групповой состав бензинов. Определение аренов в бензинах проводят, как правило, комбинированным методом анилиновых точек (ГОСТ 12329—77).
Сущность метода сводится к расчету массового содержания аренов А, %, причем исходят из изменения критических температур взаимного растворения равных объемов бензина и анилина (анилиновая точка) до и после извлечения аренов:
А = К (<2 —1\),
где К — расчетный коэффициент, характеризующий содержание аренов в данном продукте, вызывающее понижение анилиновой точки на 1°С;^и/2 — анилиновые точки исходного и деароматизированного продуктов, °С.
Значение К зависит от строения аренов и их содержания в продукте. Поэтому при анализе бензинов их необходимо предварительно разогнать (пользуясь колбой с дефлегматором) на узкие фракции: бензольную (60—95°С), толуольную (95— 122 °С), ксилольную (122—155°С) и остаточную. В каждой фракции содержание аренов определяют отдельно.
Значение К в этих фракциях изменяется следующим образом:
Фракция, °С 60-95 95—122 122—155 155—175
Массовое содержание аренов во фракции, % до 20 1,20 1,22 1,30 1,40
20—40 1,18 1,20 1,22 1,30
При анализе бензинов-растворителей с низким содержанием аренов (1,5; 3,0 и 5,0%) — значение К соответственно 1,00; 1,16; 1,17.
Содержание аренов в бензине А определяют по формуле:
A— (AtPt + А2В2 + ... + i4raBn)/100,
где АиА3, .... Ап — массовое содержание углеводородов в отдельных фракциях, %; Ви В2, ...., Вп — массовое содержание фракций в беизиие, %.
Для определения группового состава бензина методом анилиновых точек необходимо отделить арены, содержащиеся в исходном продукте. Это можно осуществить химическим методом— сульфированием 98,5—99 %-й серной кислотой или физико-химическим методом—хроматографией на силикагеле. Второй метод быстрее и проще.
Структурно-групповой состав керосиновых и масляных фракций. Имеется несколько методов анализа, позволяющих в первом приближении судить о структуре гибридных углеводородов, входящих в средние и тяжелые фракции нефти. Они основаны на изучении большого числа индивидуальных углеводородов и их смесей. Накопленный опытный материал позволил найти закономерности между распределением углерода в различных структурных фрагментах молекулы и физическими кон-стантами углеводородов и их смесей. Основанные на эмпирических расчетах, они не могут претендовать на высокую точ-. ность. Тем не менее существующие методы служат наилучшим и самым простым способом анализа указанных фракций нефти.
Метод п—р — М353 (показатель преломления — плотность — молекулярная масса). Этот метод, разработанный Бан-Несом и Ван-Вестеном в 1954 г., дает возможность находить распределение углерода и содержание колец в нефтяных фракциях, в которых нет алкенов. Метод позволяет составить представление о «средней» молекуле данной фракции, которая содержит углерод, входящий в ароматические, алициклические кольца и насыщенные алифатические соединения. Углерод, входящий в алифатические соединения, включает углерод алканов и алкильных заместителей при алициклических и ароматических кольцах. Сумма всех «видов» углерода равна 100%. Под определением числа колец подразумевается определение числа ароматических и алициклических колец в средней молекуле или в среднем во фракции.
Для получения среднестатистических значений при исполь-» зовании метода приняты следующие вполне обоснованные допущения: 1) все циклы (алициклические и ароматические) — шестичленные, 2) все кольца находятся в катоконденсирован-ном состоянии.
Для определения структурно-группового состава нефтепродукта по методу п — р — М необходимо знать: показатель преломления (с точностью до ±0,0001), плотность (с точностью до ±0,0002) и молекулярную массу (с точностью до ±3%). Расчет ведут по эмпирическим уравнениям:
Для жидких фракций (константы опреде- Для твердых фракций (константы стреляются прн 20 “С) деляются при 70 °С)
При высоком значении Са, Скол, Ка, Ко
Са = 3660/М + 430(2,51Дп — Др) СА = 3660/М + 410(2,72Ап - Др)
Скол = 10 000/М+ 820(Др—1,11Дп) CK0J, = 11 500/М + 775 (Др — 1,11Дп)
Ki = 0,44 + 0.055М (2,51 Дп — Др) Ка = 0,41 + 0,055М(2,42Дп — Ар)
1,33 + 0,146 М(Др —1,11Дп) Ко = 1,55 + 0,146 М(Др— 1,11Дл)
кол»
С А = 3660/Л1 + 670(2,51 Ап — Ар) Скол = ю 600/М + 1440(Др —
— 1,11Дп)
Ка = 0,44 + 0,80 М(2,51Дп — Др) Ко = 1,33 + 0,180 Af (Др — 1,11Дя)
Сл = 3660/М + 720 (2.42Д п — Ар) Ск„л — 12 100/Л1 + 1400 (Др —
— .1,11Дп)
КА = 0,41 + 0,080 М(2,42Дп — Др) Ко= 1,55 + 0,180 М(Др — 1,11 Дп)
Здесь Сл — массовое содержание углерода в ароматических структурах, %; СКол — массовое содержание углерода в кольчатых структурах, %; Ка — число ароматических колец в молекуле (среднее); Ко — общее число ароматических и алициклических колец в молекуле (среднее).
Высокими значениями Сд, Ск0л, Ка, Ко считаются такие, для которых алгебраическая сумма выражений в круглых скобках (Ал — Ар) положительна; если эта сумма отрицательна, следует вести расчет по формулам для низкого значения указанных показателей.
Необходимые для расчета факторы Ап и Ар представляют собой разность между соответствующими показателями нефтепродукта и гипотетического насыщенного углеводорода нормального строения:
Для жидких фракций Для твердых фракций
Дп = л» _ 1,4750 Дп = п™ — 1,4600
Др = pf _ 0,8510 Др = pf - 0,8280
Доля углерода, содержащаяся в алициклических структурах, определяется по разности.
6.3. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Идея хроматографического метода анализа — использование для разделения веществ давно известного явления избирательной сорбции — принадлежит русскому ботанику М. С. Цвету. В 1903 г. он сформулировал принцип метода и показал возможность его практического осуществления в жидкостно-адсорбционном варианте для разделения хлорофилловых пигментов листьев на составляющие с различной окраской. Отсюда происходит название метода — хроматография, т. е. цветопись. Однако сам Цвет высказал предположение, что метод применим не только к окрашенным, но и к бесцветным веществам.
Хроматография — физико-химический метод разделения и анализа, основанный на распределении компонентов между двумя фазами — неподвижной и подвижной, непрерывно протекающей через неподвижную фазу.
Виды хроматографии и методики анализа. Известно много вариантов хроматографии, которые классифицируют по различным признакам. В зависимости от природы явлений, лежащих
в основе разделения, различают адсорбционную, распределительную и осадочную хроматографию. В основе адсорбционной хроматографии — использование неодинаковой адсорбируемости разделяемых веществ на твердой поверхности адсорбента. В основе распределительной хроматографии — поглощение разделяемых соединений жидкостью, различия в растворимости, значениях коэффициентов распределения между двумя сосуществующими жидкими или жидкой и газовой фазами. В осадочной хроматографии используется явление образования нерастворимых соединений в результате химических реакций раз* деляемых веществ с реактивом — осадителем.
Наибольшее распространение получила классификация разновидностей хроматографии по признаку агрегатного состояния сосуществующих фаз (табл. 6.1).
Разделение компонентов можно осуществлять в колоннах на-садочного типа (колоночная хроматография), капиллярах, заполненных неподвижной жидкой фазой (капиллярная хроматография), на фильтровальной бумаге (бумажная хроматография), на тонком слое сорбента, нанесенном на стеклянную пластинку (тонкослойная хроматография). Разделять смеси можно при постоянной температуре и давлении или с программированием, т. е. с постепенным повышением по заданной программе температуры или давления газа-носителя. Все варианты хроматографии являются молекулярными, а жидкостно-адсорбционная хроматография может быть и ионообменной, осуществляемой при обмене ионов разделяемых компонентов с поверхностными ионами ионообменного адсорбента.
В соответствии с методикой проведения анализа различают три варианта хроматографии (рис. 6.1): фронтальный (а),про-явительный, или элюентный,. (б) и вытеснительный (в).
При фронтальном анализе смесь компонентов А + Б непре-
Таблица 6.1. Классификации методов хроматографии
по агрегатному состоянию подвижной и неподвижной фаз
|
Неподвижная фаза | Подвижная фаза | Название и принятое обозначение | Варианты хроматографии |
|
Жидкая (раствори тель) Твердая (адсорбент) | Газовая (газ-носитель) Жидкая Газовая (газ-носитель) Жидкая |
Газожидкостная (ГЖХ) Жидкость-жидкост-ная (ЖЖХ) Газовая адсорбционная (ГАХ) Жидкостно-адсорб-циониая (ЖАХ) |
Колоночная, капиллярная, с программированием температуры Колоночная, бумажная Колоночная, с программированием температуры Ионообменная, колоночная, тонкослойная, гра-диентио-элюентная |
Рис. 6.1. Схемы хроматографического анализа: а 6 в
а — фронтальный вариант; б — проявительный (элю-ентный) вариант; в — вытеснительный вариант
А+Б (А+Б)+В (А+БНД
рывно пропускают через хроматографическую колонку с сорбентом до тех пор, пока не выйдет слабосорбирую-щийся компонент Б; затем из колонки начинает выходить смесь компонентов. Метод не нашел широкого применения, так как он не дает полного разделения: в чистом виде выделяется только наиболее слабо адсорбирующийся компонент.
Д
А+В
А+Б
Б + В
А + Б
При проявительном (элюентном) анализе в колонку вводят определенное количество смеси А + Б и проявитель (растворитель или газ-носитель) В, сорбирующийся слабее, чем компоненты смеси. Происходит смещение зон компонента Б относительно А и разделение зон. Вариант получил наиболее широкое применение; при правильном выборе условий этот метод позволяет разделить все компоненты и проанализировать смесь.
При вытеснительном анализе в колонку вводят смесь А + Б, а затем вытеснитель Д, сорбирующийся сильнее всех компонентов. При этом методе можно получить некоторое количество чистых компонентов А и Б, но полного их разделения не достигается из-за взаимной диффузии на границе зон.
Газожидкостная хроматография. Газожидкостная хроматография, открытая в 1952 г. А. Джеймсом и А. Мартином, наиболее широко применяется в нефтехимии и нефтепереработке по сравнению с другими вариантами хроматографии, а также со всеми прочими физико-химическими и физическими методами анализа. Это обусловлено следующими преимуществами метода:
1) высокая разделяющая способность—ни один другой метод не позволяет так быстро (в течение 0,5—1 ч) проанализировать фракции нефти, состоящие из десятков и сотен компонентов; предельная эффективность колонок, достигнутая в ГЖХ, составляет приблизительно 108 теоретических тарелок;
2) высокая чувствительность — метод позволяет определять микропримеси с концентрацией до 10~10%; чувствительность детектирования в газах на несколько порядков выше, чем в жидкостной хроматографии;
3) быстрота анализа — скорость диффузии в газах приблизительно в 1000 раз выше, чем в жидкостях, поэтому в колонке быстро устанавливается равновесие и достигается высокая удельная эффективность;
4) малый размер пробы, необходимый для анализа (десятые доли миллиграмма);
5) достаточно высокая точность анализа — средняя относительная погрешность измерения концентраций 5%, а на хроматографах высокого класса с более тщательной стабилизацией основных параметров 2 % (отн.);
6) сравнительная простота аппаратурного оформления.
Принципиальная схема газового хроматографа приведена
на рис. 6.2. При ГЖХ хроматографическую колонку заполняют неподвижной фазой — инертным измельченным твердым носителем, пропитанным растворителем. Через термостатированную колонку с определенной скоростью пропускают поток газа-носителя, в который вводят с помощью микрошприца анализируемую пробу. Анализируемая смесь испаряется в испарителе, нагретом до температуры выше конца кипения фракции, и затем разделяется в хроматографической колонке.
Выходящий из колонки поток газа-носителя, содержащий пары разделенных компонентов смеси, проходит через одну из камер детектора. Через камеру сравнения детектора пропускается чистый газ-носитель. Принцип действия детекторов может быть различным. Например, в катарометрах, достаточно широко применяющихся в качестве детекторов в газовой хроматографии, используют различия в теплопроводности газа-носителя и анализируемых компонентов. Различие теплопроводности газовой среды в камерах катаромегра при прохождении через одну из них компонента смеси приводит к возникновению разности температур и электрических сопротивлений нитей накаливания, находящихся внутри камер, и в результате — раз-балансированию моста Уитстона, сигнал катарометра усиливается потенциометром и регистрируется самописцем на хроматограмме в виде пика соответствующего компонента.
Ввод про5ы
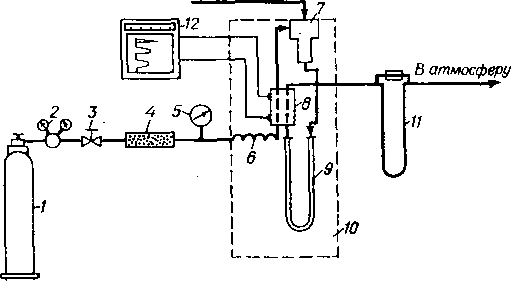
Рнс. 6.2. Принципиальная схема газового хроматографа:
/ — баллон с газом-носитслсм; 2 — редуктор; 3 ~ пентиль тонкой регулироики; 4 — осушитель; 5 —манометр; в — подогреватель; 7 — узел виода пробы; 8 — детектор; S — хроматографическая колонка; 10 — термостат; // — измеритель скорости потока; /2 —электронный потенциометр
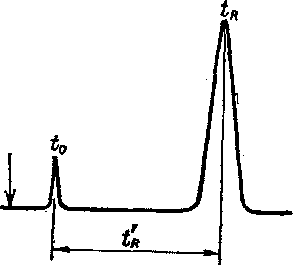
Рис. 6.3. Расчет времени удерживания компонентов при хроматографическом анализе
Широко распространены в газовой хроматографии также пламенно-ионизационные детекторы, отличающиеся более высокой чувствительностью по сравнению с катарометрами. Иногда используются и специальные детекторы (электронозахватный, микрокулонометрический, инфракрасный и т. п.), высокоселективные по отношению к определенным группам соединений. В конце 80-х годов в практику введены атомно-эмиссионные детекторы, селективные при анализе элементов, например, серосодержащих компонентов нефтяных фракций.
В ГЖХ используют различия в летучести компонентов смеси, в геометрической структуре их молекул и интенсивности взаимодействия с неподвижной фазой. Селективные неподвижные фазы обеспечивают различную растворяющую способность по отношению к анализируемым веществам и взаимное смещение зон компонентов смеси. Различают селективность как сро-собность к разделению каких-либо двух компонентов, групповую селективность как способность к разделению компонентов двух гомологических рядов, например алканов и аренов, а также селективность по молекулярным массам — способность к разделению компонентов одного гомологического ряда. Как и в процессах экстракции, экстрактивной и азеотропной ректификации," абсорбции, селективность растворителей в ГЖХ можно характеризовать отношением коэффициентов активности разделяемых компонентов й растворителе. Значения коэффициентов активности связаны с параметрами удерживания компонентов в хроматографической колонке.
Время от момента пуска пробы в колонку до выхода максимума пика называется временем удерживания tg (рис. 6.3). Оно складывается из времени пребывания компонента в газовой фазе 4о и времени, когда молекулы находятся в сорбированном состоянии, tR. Значение t0 зависит от доли пустот в заполненной колонке («мертвого объема»). Оно может быть определено по времени удерживания практически несорбирую-щихся веществ, например воздуха. Истинная удерживающая способность колонки характеризуется исправленным временем удерживания:
tR==t^~ to
Время удерживания соединений на данной неподвижной фазе зависит от условий хроматографического анализа: скорости газа-носителя, количества растворителя в колонке. Для сравнения удерживания различных соединений на одной и той же неподвижной фазе или одного и того же вещества на различных неподвижных фазах «асто используют значения удельных удерживаемых объе., Vg. Удельный удерживаемый объем — это объем газа-носи^ля, приведенный к нормальным условиям и отнесенный к 1 г растворителя в колонке, который надо пропустить, чтобы элюировать данное вещество:
Ft'R 273,15 Vg=——/•
* ш Т
Здесь F — объемная скорость газа-носителя; <о — масса растворителя в колонке; Т — температура измерителя скорости потока газа-иосителя, К; / — поправка, учитывающая сжимаемость газа-иосителя в колонке и рассчитываемая по формуле:
i — 1 (Р./Р^)2-!
1 2 (Р,/Р2)3- 1 ’
где Рь Рг — давление соответственно на входе в колонку и выходе из нее.
Зная удельные удерживаемые объемы, можно рассчитать коэффициенты активности разделяемых компонентов в растворителе при состоянии, близком к бесконечному разбавлению, и оценить селективность данной неподвижной фазы:
Y? = 273,15R/MVgPl
где R — универсальная газовая постоянная; М — молекулярная масса растворителя; Р°—давление насыщенного пара компонента при температуре колонки.
Для идентификации компонентов смесей широко используют относительные параметры удерживания, в частности относительное время удерживания:
^отн ^/?Аст’
где — исправленное время удерживания стандартного вещества (чаще
Г
всего какого-либо к-алкаиа), определенное при тех же условиях, что и tg.
В качестве относительного параметра для идентификации широко используют также индексы Ковача, рассчитываемые по формуле:
If? — Ig tn
Ig*n+r — Ig'n
где tn, tn+i — исправленные рремена удерживания «-алкаиов с числом атомов углерода лил+1.
Сущность системы Ковача состоит в том, что время удерживания данного соединения сопоставляется с временем удерживания к-алканов, значения индексов удерживания которых приняты равными числу углеродных атомов, умноженному на 100. При расчете индекса Ковача желательно подобрать алканы так, чтобы идентифицируемое соединение элюировалось между ними.
Значения относительных времен удерживания и индексов Ковача различных веществ, в том числе углеводородов, на многих типичных неподвижных фазах приведены в справочной литературе. Сопоставляя относительные характеристики удерживания компонентов смеси с литературными данными, проводят идентификацию. При наличии предполагаемого вещества в чистом виде некоторое количество его добавляют к анализируемой смеси и наблюдают за изменением высоты и формы пика. Если пик принадлежит добавляемому соединению, то его высота должна увеличиться, а ширина на половине высоты остаться неизменной. Для повышения достоверности идентификации аналогичный прием повторяют, используя колонку с другой неподвижной фазой,1 отличающейся по полярности от первой.
При отсутствии эталонов или эталонных смесей для идентификации можно использовать линейные зависимости между величинами lg Vg (или индексов удерживания) и такими характеристиками анализируемых веществ, как число углеродных атомов в молекуле, температура кипения, логарифм давления насыщенного пара. Эти зависимости, как правило, достаточно хорошо выполняются для соединений одного гомологического ряда.
Для идентификации сложных смесей, нестабильных веществ, практически нелетучих высокомолекулярных соединений часто используют аналитическую реакционную газовую хроматографию — вариант, в котором хроматографический и химический анализ сочетаются в единой хроматографической схеме. Задача метода состоит в том, чтобы в результате химических реакций получить новую смесь, компоненты которой разделяются или идентифицируются лучше, чем компоненты исходной смеси. Широкое применение при этом находит метод вычитания, при котором проводят два хроматографических анализа — исходной смеси до и после поглощения определенной группы компонентов. Таким способом можно, 'например, устанавливать наличие во фракциях непредельных углеводородов, селективно поглощая их в реакторе с силикагелем, обработанным серной кислотой. При реакционной газовой хроматографии используются также реакции гидрирования, дегидрирования, этерификации (для анализа карбоновых кислот в вйде эфиров), пиролиза высокомолекулярных соединений.
Широко применяется и хромато-масс-спектромет-рия — хроматографическое разделение смеси и идентификация компонентов по масс-спектрам.
В ряде случаев индивидуальные компоненты выделяют препаративной хроматографией и идентифицируют спектральными или другими независимыми методами.
Идентификация отдельных групп соединений возможна с помощью специальных детекторов, имеющих повышенную чувствительность к данным соединениям. Так, кулонометрический детектор, действие которого основано на титровании продуктов сгорания элюата электролитическим бромом, может использоваться для анализа серосодержащих соединений. Электронозахватный детектор имеет высокую чувствительность к фосфор- и галогенсодержащим соединениям, обладающим большим сродством к электрону.
Хроматографические методы позволяют проводить не только идентификацию, но и количественный анализ. Состав смеси можно рассчитать по площадям пиков, которые определяют при помощи интеграторов, планиметров, взвешиванием вырезанных пиков или рассчитывают как произведение высоты пика на его ширину на половине высоты. При узких или не полностью разделяющихся пиках меньшую погрешность при расчете состава дает использование вместо площадей пиков пропорциональных им значений произведений высот пиков на время или удельный удерживаемый объем.
В связи с тем что чувствительность детекторов к различным соединениям неодинакова, при количественном анализё смесей необходимо учитывать поправочные коэффициенты. При этом можно использовать несколько методов.
Метод нормализации основан на том, что сумма площадей 2)5,- всех пиков с учетом поправочных коэффициентов принимается за 100%. Калибровочные коэффициенты К определяют, анализируя стандартную смесь известного состава, состоящую из тех же компонентов, что и анализируемая смесь. Для одного из компонентов принимают Ki — I и рассчитывают поправочные коэффициенты для остальных компонентов. Состав анализируемой смеси рассчитывают по формуле:
Метод можно использовать лишь в случае, если все компоненты смеси регистрируются на хроматограмме.
Метод внутренней нормализации удобнее всего использовать, если не все компоненты смеси регистрируются на хроматограмме или необходимо определить содержание лишь одного или нескольких компонентов в смеси. Метод основан на добавлении к анализируемым компонентам известных количеств вещества, выбранного в качестве внутреннего стандарта (или метки). Для калибровки проводят хроматографический анализ ряда смесей стандарта с каждым из анализируемых компонентов при различных их соотношениях. Затем известное количество вещества, выбранного в качестве внутреннего стандарта, добавляют к анализируемой пробе и, определив соотношение площадей пиков искомого компонента и стандарта, по калибровочному графику рассчитывают концентрацию компонента в смеси.
Метод абсолютной калибровки можно применять при анализе газовых смесей. В этом случае в колонку дозируют определенные количества компонентов qi, измеряют площади пиков Si и строят калибровочный график Si = f(qi). Дозируя затем известное количество смеси в колонку и пользуясь калибровочным графиком, рассчитывают содержание компонента в смеси., Способ применяется редко из-за погрешностей при дозировании микрошприцем (особенно велики погрешности при дозировании жидкостей) и необходимости строго постоянного режима работы хроматографа при калибровке и анализе.
При методах внутренней нормализации и нормализации не требуется знать количество пробы, введенной в колонку.
Капиллярная хроматография, открытая в 1957 г. М. Дж. Голеем, значительно расширила аналитические возможности хроматографии, в частности при исследовании индивидуального состава нефтяных фракций. Капиллярные колонки — это металлические или стеклянные свернутые в спираль капилляры внутренним диаметром около 0,25 мм и длиной в несколько десятков метров, заполненные неподвижной фазой — растворителем.
Благодаря большой длине капиллярные колонки значительно более эффективны, чем обычные набивные, заполненные твердым носителем, пропитанным растворителем, длина которых составляет несколько метров. Эффективность капиллярных колонок составляет до 3000—5000 теоретических тарелок на 1 м, т. е. при длине 200 м эффективность может достигать 106 теоретических тарелок. Такие колонки успешно используют для разделения соединений с очень близкими летучестями, в том числе при анализе изотопов и изомеров.
При использовании набивных колонок даже анализ изомеров углеводородов Се представлял определенные трудности, а капиллярные колонки с неполярной неподвижной фазой — скваланом — позволяют анализировать все изомеры не только гексана, но и гептана, Октана. Применение капиллярных колонок позволило провести почти полную идентификацию компонентов бензиновых фракций нефтей, перегоняющихся до 175°С. Присутствующие в этих фракциях алкилбензолы можно анализировать после предварительного их выделения жидкостной адсорбционной хроматографией, экстракцией или без предварительного выделения, непосредственно в исходной фракции — на колонках с высокоселективными неподвижными фазами. Например, на полиэтиленгликоле (ПЭГ-600) индекс удерживания бензола при 100 °С равен 988, т. е. бензольный пик на хроматограмме выходит между нонаном и деканом. На еще более высокоселективной фазе—Ы,Ы'-бис(2-цианоэтил)формамиде индекс удерживания бензола при 180°С равен 1800, т. е. бензол удерживается так же, как октадекан.
Промежуточное положение между обычными набивными и капиллярными колонками занимают микронабивные колонки, имеющие внутренний диаметр 0,8—1 мм. Эффективность микро-набивных колонок на единицу длины выше, чем капиллярных, за счет меньшей доли пустот в колонке. Микронабивные колонки эффективнее и обычных набивных с диаметром в несколько миллиметров, так как в них меньшую роль играют поперечная диффузия и стеночный эффект, йриводящие к размыванию хроматографических полос.
Анализ прямогонных бензиновых фракций проводят методом ГЖХ с использованием капиллярных колонок. Разработанные методики анализа бензинов можно условш} разделить на две группы. К первой относятся методы, использующие для идентификации индексы удерживания Ковача, ко второй — методы, основу которых составляет порядок выхода углеводородов на стандартной, обычно неполярной фазе (сква-лане) при строго заданных рабочих температурах.
Рекомендуется предварительно разделять бензиновую фракцию ректификацией, отбирая фракции н. к.— 125°С и 125— 150°С. Бензиновые фракции нефтей и конденсатов представляют собой сложные смеси углеводородов различного строения: до 125°С выкипают 70 компонентов, а в интервале 125— 150 °С — 130 компонентов.
Хроматографирование проводят на капиллярной колонке длиной 50—100 ад и внутренним диаметром 0,2—0,3 мм, заполненной скваланом. Анализ фракций н. к.— 125°С осуществляется при двух температурах, оптимальными являются 50 и 80 °С. Пример хроматограммы фракции н. к. — 125°С нефти Самотлорского месторождения приведен на рис. 6.4. Селективность неподвижной фазы зависит от температуры, поэтому при другой температуре (80°С) порядок выхода компонентов несколько меняется. Пики, представляющие собой дуплеты или триплеты, при другой температуре соответствуют индивидуальным углеводородам или выходят в других комбинациях. Например, при 50 °С н-октан выходит вместе с транс,транс,транс-1,2,3,4-тетраметилциклопентаном (пик 47) и не полностью отделяется от них транс- 1,2-диметилциклогексан (пик 48), а при 80 °С они разделяются.
Качественную расшифровку хроматограмм лучше всего на-
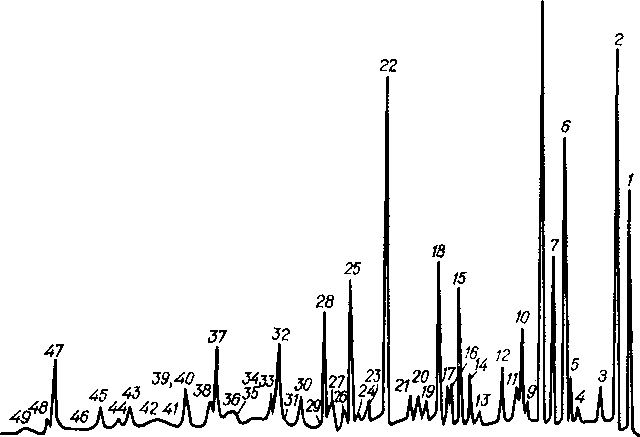
w-гептан, w-октан. Возросшие на хроматограмме пики относятся к этим углеводородам.
Для идентификации циклоалканов и аренов достаточно знать порядок их выхода на хроматограммах при соответствующей температуре.
Количественную расшифровку хроматограмм проводят методом внутренней нормализации с измерением высоты пиков h и расстояния максимума пика от момента ввода пробы I. Содержание каждого компонента на исследуемую фракцию рассчитывают как отношение Iti/EUti).
Фракцию 125—150 °С предварительно разделяют жидкостной хроматографией на силикагеле на ареновую и алкан-цикло-алкановую часть, которые затем порознь анализируют на капиллярной колонке.
Арены хорошо разделяются на сквалане при 80°С, выходя в порядке повышения температуры кипения. Хроматограммы алкан-циклоалкановой части снимают при двух температурах (80 и 106°С) или в режиме линейного программирования температуры (например, при начальной температуре анализа 50°С и скорости подъема температуры 1°С/мин).
Анализ высококипящих компонентов, входящих в состав керосино-газойлевых и масляных фракций нефти,— значительно более сложная задача по сравнению с анализом бензиновых фракций. Полная идентификация даже углеводородов керосиновых фракций — практически невыполнимая задача. Однако метод ГЖХ позволяет получать данные об индивидуальном составе отдельных групп углеводородов, предварительно выделенных из нефтяных фракций — н-алканов, углеводородов изопреноидного строения алкиладамантанов, аренов.
Для анализа высококипящих компонентов нефти методом ГЖХ необходимо использовать термостабильные неподвижные фазы, практически нелетучие при температуре анализа. Высокой термостабильностью характеризуются силоксановые неподвижные фазы. Например, верхний температурный предел применимости диметилсиликоновой жидкости OV-lOl, имеющей формулу [(CH3)2SiO]„, составляет 325—375°С. Фазы типа OV* как правило, неполярны или слабополярны.
Новая группа неподвижных фаз типа силара представляет собой полимеры, содержащие фенильные и цианалкильные функциональные группы, сшитые между собой полисилоксано-выми цепочками. Эти фазы обладают высокой полярностью и позволяют обеспечить селективность при высоких температурах (до 275°С), в этих условиях другие фазы становятся малоселективными.
Наиболее высокотемпературные фазы типа дексила, имеющие карборансилоксановую структуру, были предложены в 70-х годах. Так, формула дексила-300 — полимера со средней
“ СНз СНз СНз СНз “
I I 1 1
—S i—С В, оН Г оС—S i—О—S i—О—S i—О—
_ СНз СНз СНз. СНз
Интервал рабочих температур для этой неполярной малоселективной фазы 50—450 °С. Известны и более полярные кар-бор ансилоксановые фазы, содержащие метилфенилсилоксано-
вые или метилцианэтильные группы, с максимальной рабочей температурой использования 400 °С.
Применение фаз типа дексила позволило анализировать углеводороды и гетерогенные соединения нефти молекулярной массой выше 800 с температурой кипения на 100—150°С выше по сравнению с фракциями, анализировавшимися методом
ГЖХ ранее. Так, удалось проанализировать «-алканы до СввУ полициклические арены до коронена включительно.
Газовая адсорбционная хроматография. Большое распространение ГЖХ по сравнению с газовой адсорбционной хроматографией обусловлено широким выбором различных по селективности неподвижных жидкостей, создающим большие возможности для анализа разнообразных смесей. Кроме того, благодаря однородности жидкостей изотермы растворимости практически линейны и пики анализируемых соединений, как правило, симметричны. Выбор же адсорбентов ограничен и они неоднородны, что приводит к нелинейности изотерм адсорбции, размыванию и несимметричности пиков, ухудшению.разделения..
Однако и ГЖХ не свободна от недостатков: летучесть и нестабильность неподвижных фаз затрудняет анализ микропримесей, а также высокомолекулярных соединений при высоких температурах; слабая растворимость газов в жидкостях и слишком малое время удерживания затрудняют анализ низкокипя-щих соединений.
Газовая адсорбционная хроматография (ГАХ) отличается большей термической стабильностью неподвижных фаз—адсорбентов— и может успешно применяться как при высоких температурах для анализа высококипящих соединений, так и при низких — для анализа природных и нефтяных газов. Для анализа слабоадсорбирующихся молекул газов и легкокипящих углеводородов используют адсорбенты с большой удельной поверхностью— цеолиты, тонкопористые силикагели. По мере увеличения объема анализируемых молекул необходимо применять всс более макропористые адсорбенты с менее развитой поверхностью. Выпуск однородных адсорбентов, в частности цеолитов >1 пористых полимеров, так называемых порапаков, па основе сополимеров стирола, этнлетирола, дивинилбензола, N-винил-пирролидона, позволил уменьшить несимметричность пиков и расширить область применения ГАХ.
Интересным адсорбентом для ГАХ является графитирован-ная сажа. Адсорбция на ней осуществляется за счет неспецифических дисперсионных сил, и при разделении смесей определяющую роль играет число контактов звеньев молекулы с плоской поверхностью частиц сажи. Например, время удерживания углеводородов С6 в соответствии с уменьшением поверхности контакта изменяется в следующем ряду: гексан >
> бензол > циклогексан. Графитированную сажу применяют и для анализа изомеров и изотопов.
Перспективным новым адсорбентом является карбосфер (сферокарб) — углеродный адсорбент типа молекулярных сит с размером пор около 1,5 нм. На нем быстро элюируется вода, разделяются азот и кислород.
Анализ нефтяных газов может быть проведен методом ГАХ в системе нз двух колонок. Первая колонка с цеолитом СаХ служит для определения содержания неуглеводородных компонентов и низкокипящих углеводородов, элюирующихся в следующем порядке: Н2, 02, N2, СН4, СО, С2,Н6, СзН8, С02, С2Н4. Анализ проводят в режиме программирования температур. Вторая колЬнка содержит в качестве адсорбента трепел Зикеевского карьера (ТЗК), модифицированный вазелиновым маслом. На этой неподвижной фазе анализируют углеводороды С2 — С5, в том числе цис- и гранс-изомеры, алкадиены, алкины. ТЗК — единственный адсорбент, на котором, не применяя низких температур, можно отделять изобутены от бутенов.
Жидкостная адсорбционная хроматография. Жидкостная адсорбционная хроматография применяется для группового разделения углеводородов на алкано-циклоалкановую и ареновую фракции, а также для разделения аренов по степени цикличности. Хроматографические колонки заполняют силикагелем или двойным адсорбентом — оксидом алюминия и силикагелем. В качестве десорбентов при анализе керосиновых и масляных фракций для вымывания насыщенных углеводородов используют н-алканы С5 — С7, для десорбции ароматических и гете-роатомных компонентов — бензол, спиртобензольные смеси, ацетон, хлороформ. Применение ступенчатого или непрерывного увеличения полярности подвижной фазы позволяет значительно уменьшить время удерживания веществ. Этот метод называется градиентным элюированием.
Пробу хроматографируют, разделяют на хроматографические фракции, определяют выход каждой фракции после от-
20
гона растворитслси, показатель преломления hd, дисперсию и
строят хроматограмму пд по оси ординат — выход фракции по оси абсцисс. Хроматограмма помогает сгруппировать соседние
фракции. Фракцию до резкого подъема кривой по относят к алкано-циклоалкановой. Границу между алкано-циклоалка-нами и аренами можно определять и по возрастанию дисперсии, а также по резкому увеличению объемов растворителя, пошедшего на десорбцию микрофракций.
Начало элюирования аренов можно устанавливать с помощью формолитовой реакции — по образованию комплексов с формалином в сернокислотной среде.
Проводя жидкостное адсорбционное хроматографирование, алкано-циклоалканов (для фракций с началом кипения «250°С), можно разделить углеводороды по следующим подгруппам: 1) нормальные или слаборазветвленные алканы, застывающие при температуре выше 20°С; 2) разветвленные алканы изостроения (лд = 1,45— 1,47); 3) моноциклические
2о •
циклоалканы (лD — 1,47— 1,48); 4) бициклические циклоал-20
каны (л?> = 1,48— 1,49); 5) три- и полициклические'циклоалканы (до резкого увеличения объема растворителя при десорбции). Арены можно также подразделить на легкие, в основном 20 20 моноциклические (hd <. 1,53), средние—бициклические (ли =
20
= 1,53— 1,55) и тяжелые — три- и полициклические (лд >
> 1,55). После тяжелых аренов иногда наблюдается понижение показателя преломления и затем выделяются смолистые вещества.
Широкое распространение, в особенности за рубежом, при групповом анализе углеводородных смесей получил метод жидкостной хроматографии на силикагеле в присутствии флуоресцирующих (люмицесцирующих) индикаторов — метод ФИ А. В колонку с силикагелем вводят анализируемую фракцию с небольшим количеством флуоресцирующих индикаторов и красителя. «Ароматический» индикатор хорошо растворим в аренах, но не растворяется в других углеводородах. При ультрафиолетовом облучении колонки зона аренов дает ярко-голубую флуоресценцию. Найдены также «олефиновые» индикаторы, растворимые в алкенах и вызывающие флуоресценцию в УФ-свете алкеновой зоны хроматографической колонки. По отношению высоты соответствующей зоны к высоте слоя адсорбента рассчитывают содержание алкенов и аренов в нефтяной фракции или нефтепродукте.
Жидкость-жидкостная хроматография. В жидкость-жидкост-ной хроматографии (ЖЖХ) молекулы образца распределяются между жидкими неподвижной и подвижной фазами (подобно жидкостной экстракции), которые не должны растворяться друг в друге.
Жидкость-жидкостная хроматография при исследовании химического состава нефтей применяется ограниченно. Ее можно
проводить в колонках с носителем, пропитанным растворителем, или на бумаге. Так, методом ЖЖХ возможно концентрирование алканов из смесей с моно- и бициклическими цикло-алканами бензиновых фракций при использовании в качестве неподвижной фазы анилина или метилового эфира этиленгли-коля на силикагеле, а в качестве подвижной фазы — перфтор-алифатических соединений, обладающих повышенной растворяющей способностью по отношению к алканам.
В последнее время наблюдается возрождение ЖЖХ благодаря созданию совершенных жидкофазных хроматографов с чувствительными детекторами и автоматической записью хроматограмм. Для повышения скорости анализа и эффективности разделения ЖЖХ проводят под давлением до 30 МПа. Наиболее целесообразно использование ЖЖХ для исследования высокомолекулярных соединений нефти.
Бумажная хроматография, открытая в 1941 г. А. Мартином и Р. Синджем, является одним из вариантов ЖЖХ. Роль хроматографической колонки выполняет полоска пористой бумаги, неподвижной фазой служит вода, удерживаемая волокнами целлюлозы, а подвижной — органические растворители. Бумажная хроматография применяется при анализе смолистых веществ и асфальтенов. Полоску бумаги погружают в спиртобензольный раствор образца и оставляют на 12—14 ч, в течение которых на бумаге образуется хроматограмма, а растворитель улетучивается. При облучении бумаги ультрафиолетовым светом зона смол дает ярко-желтую люминесценцию, а асфальтены — темно-коричневую.
Вместо бумажной хроматографии можно использовать тонкослойную хроматографию. Адсорбент, например силикагель, распределяют равномерным слоем толщиной «1 мм на стеклянной пластине, для закрепления слоя добавляют инертное вяжущее вещество. Анализируемый образец наносят на один край пластины и погружают ее в растворитель, который постепенно мигрирует в слое адсорбента. При этом происходит образование зон компонентов образца, причем как и в хроматографической колонке, быстрее всего перемещаются наименее полярные компоненты. Методом тонкослойной хроматографии недавно было установлено высокое содержание (до 15—20%) алкенов в некоторых нефтях.
Г е л ь - х р о м а т о гр а ф и я, или э к с к л ю з и о н н а я х р о м а то г р а ф и я — еще один вариант жидкостной хроматографии, при котором молекулы разделяемой пробы элюируют в зависимости от их объема и формы. Заполнитель колонки (гель) имеет поры определенного размера. Если в разделяемом образце есть молекулы, размеры которых не позволяют им проникать в поры геля, то они проходят с потоком элюента только между частицами геля и быстро выходят из колонки.
Молекулы небольшого размера могут проникать во все поры геля, путь их удлиняется, и они задерживаются в колонке дольше других компонентов. Молекулы средних размеров проникают только в некоторые поры, путь их оказывается средним по длине.
Гель-хроматография применяется для анализа тяжелых нефтяных остатков, кипящих при температурах выше 400°С, котельных топлив, для анализа которых другие методы непригодны. В 1965 г. К. Альтгельт обнаружил возможность фракционирования асфальтенов методом гель-хроматографии. Разделение на фракции по молекулярной массе тяжелых нефтепродуктов, прежде всего битумов, позволяет получать более надежные данные при последующем исследовании их другими методами, например ЯМР.
В качестве колоночной насадки в большинстве случаев применяют сефадекс LH-20 и стиролдивинилбензольные гели, через которые алканы и циклоалканы элюируются растворителями по молекулярно-ситовому механизму. Порядок элюирования полициклических аренов зависит от применяемого растворителя. При использовании хлороформа, тетрагидрофурана и для аренов сохраняется порядок, типичный для гель-фильтрации. Однако при элюировании кетонами, спиртами, ацетонитрилом может проявляться адсорбционный эффект, вследствие которого с увеличением числа ароматических колец время удерживания соединений увеличивается.
Гель-хроматография не получила широкого применения из-за трудности надежной интерпретации результатов разделения. Тем не менее метод перспективен: внедрение его дает возможность вести контроль за изменении состава по молекулярным массам в процессах нефтепереработки, определять содержание отдельных фракций в нефтях, оценивать качество нефтепродуктов, идентифицировать сырые нефти, контролировать загрязнение окружающей среды нефтепродуктами.
Препаративная хроматография благодаря высокой разделяющей способности колонок и использованию селективных неподвижных фаз позволяет разделить практически любые смеси, н том числе азеотропы и изомеры. Для выделения веществ с целью последующей идентификации другими методами можно пользоваться препаративными приставками к обычным хроматографам с колонками диаметром до 20 мм и производительностью несколько десятков граммов вещества в сутки. Для выделении соединений с целью исследования их свойств или использования в лабораторных синтезах применяют специальные препаративные хроматографы с колонками диаметром 100—200 мм и производительностью 1 кг в сутки и более. Для получения реагентов промышленного синтеза используют производственную хроматографию — колонны диаметром 1—3 м, имеющие производительность до 1000 т/год. Так, разработаны* хроматографические колонны диаметром 52—120 см для производства 100—1200 т/год тиофена, толуола и индола.
Основные преимущества хроматографии перед ректификацией заключаются в меньших энергетических затратах при низких значениях коэффициентов относительной летучести разделяемых ключевых компонентов, отсутствии большого числа колонн и возможности селективного удаления примесей за одну операцию.
Основной недостаток препаративной хроматографии — сравнительно низкая производительность. Увеличение диаметра колонок приводит к снижению эффективности разделения из-за-стеночного эффекта: плотность неподвижной фазы у стенок: колонки при их набивке всегда меньше, чем в центре. Поэтому доля пустот и скорость потока у стенок больше, чем в центре, что приводит к размыванию хроматографических полос.
Повышение эффективности разделения возможно при применении циркуляционной хроматографии, позволяющей осуществить препаративное разделение смесей с коэффициентом относительной летучести а =1,013—1,10, например разделение смеси этилбензола и л-ксилола.
Для повышения производительности возможно вместо обычного периодического процесса, при котором в каждый момент времени в разделении принимает участие только часть сорбента, применение непрерывной хроматографии с противоточ-ным движением сорбента и подвижной фазы. Наиболее перспективен вариант, в котором слой сорбента неподвижен относительно стенок вращающейся кольцевой колонны, а газ-носи-тель можно вводить в различные точки колонны.
Увеличение производительности достигается и при применении нового метода — хр о м а д ис т и л л я ц и и, различные варианты которого предложены А. А. Жуховицким с сотрудниками. Этот метод находится на стыке хроматографии и ректификации, когда хроматография осуществляется с использованием в качестве неподвижной фазы компонентов разделяемой смеси. В трубку с инертным наполнителем— стеклянными или металлическими шариками — вводят разделяемую смесь и пропускают газ-носитель. При этом на заднем фронте жидкости происходит испарение, а на переднем при охлаждении обеспечивается процесс конденсации.
Метод можно использовать в аналитических и препаративных целях, а также для получения кривых истинных температур кипения (НТК) нефтяных фракций. Преимущества хрома-дистилляции перед ректификацией в последнем случае—более четкое разделение, вплоть до полного разделения компонентов,, значительно меньшие объемы пробы (150 мкл) для анализа,, более низкая температура анализа, что позволяет получать дан--«ые о фракционном составе более высококипящих нефтепродуктов.
Хромадистилляционная разгонка проводится на колонке с отрицательным температурным градиентом в режиме линейного программирования температуры и дает распределение узких фракций до 650 °С. Для построения кривой ИТК используют калибровочные графики в координатах h — (Тк/ТоП) (h — сигнал детектора, соответствующий давлению насыщенного пара «-алканов при температуре опыта ГоП; Тк — нормальная температура кипения н-алканов). Расхождение между двумя методами (хромадистилляционным и стандартным ректификационным на аппарате АРН-2) составляет 3—5°С.
в.4. МАСС-СПЕКТРОМЕТРИЯ И ХРОМАТО-МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометр.и я впервые была использована для анализа легкокипящих нефтяных фракций в 1940 г. После появления в 1959 г. масс-спектрометров высокого разрешения, обеспечивающих разделение углеводородных и гетероатомных ионов с близкими массами, и создания систем прямого ввода образца в ионный источник оказалось возможным использовать этот метод и для анализа средних и тяжелых нефтяных фракций. Современный этап развития масс-спектрометрии характеризуется разнообразием способов ионизации вещества, быстродействием, сочетанием с газовой хроматографией, полной автоматизацией эксперимента и обработкой результатов с помощью ЭВМ.
Масс-спектрометр содержит следующие основные узлы: источник ионов, в котором происходит ионизация молекул анализируемого вещества; анализатор, осуществляющий разделение ионов; систему ввода вещества в ионный источник; систему регистрации масс-спектра; систему откачки, обеспечивающую необходимый вакуум.
Исследуемую фракцию в газообразном состоянии из баллона напуска 1 (рис. 6.5) подают через молекулярный иатекатель 2
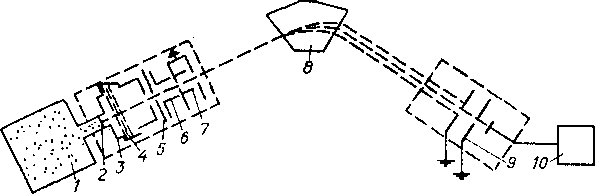
Рис. 6.5. Принципиальная схема масс-спектрометра
Обозначения см. в текст*
в камеру 3. Ионизация и диссоциация молекул исследуемых: веществ происходят в результате электронного удара. Поток ионизирующих электронов 4 испускается накаленным катодом. Притягиваясь к аноду, электроны приобретают кинетическую энергию, достаточную для ионизации молекул. Образовавшиеся положительно заряженные ионы вытягиваются из зоны ионизации, формируются и ускоряются в электронно-оптической системе, состоящей из вытягивающей 5, фокусирующей 6 w ускоряющей 7 линз. Далее ионы движутся в магнитном поле-электромагнита & по круговым траекториям, радиус кривизны которых зависит от отношения массы иона к его заряду (т/е). При соответствующей напряженности электрического и магнитного полей в щель коллектора 9 попадают ионы с определенным значением т/е. При изменении напряженности магнитного поля или ускоряющего потенциала и остальные ионы могуг быть сфокусированы на щель коллектора. Ионы нейтрализуются на коллекторе и создают в его цепи ток, усиливаемый электрометрическим усилителем 10 и регистрируемый. Для записи масс-спектров используют электронные потенциометры.
Образование ионов, фокусировку ионного пучка и разделение ионов по массам осуществляют в условиях высокого вакуума, когда длины свободных пробегов ионов и молекул превышают размеры анализатора. Это дает возможность избежать вторичных соударений частиц, искажающих первоначальный состав и форму ионного пучка.
Могут использоваться и другие методы ионизации — химическая ионизация при столкновениях молекул анализируемого' вещества с ионами или метастабильными возбужденными атомами газа-реактанта (СН4, NH3 и др.); полевая ионизация в сильном неоднородном электрическом поле, создаваемом специальным электродом; лазерная десорбция и т. д. Однако* классические методы ионизации электронным ударом при высоких (70 эВ) и низких (10—13 эВ) энергиях электронов остаются наиболее распространенными. Энергия электронов превышает потенциал ионизации углеводородов, составляющий для алканов 10—13, алкенов 9—10, алкилбензольных углеводородов 8,5—9,5, и полициклических аренов—менее 8 эВ. Поэтому при столкновении с электронами молекулы углеводородов ионизируются, т. е. происходит отрыв валенных электронов и образование молекулярных ионов М.
Молекулярный ион диссоциирует через состояние активированного комплекса, распад которого идет преимущественно в направлении образования стабильных продуктов. Ионизация молекул протекает быстро (за 10~15 с), а распад — сравнительно длительный акт продолжительностью 10~б—10-10 с. За этот промежуток времени избыточная энергия, полученная ионизированной молекулой от электрона (сверх потенциала
Рис. 6.6. Кривые распределения интенсивностей пнков относительно ; полного ионного тока по числу атомов углерода в нонах:
I — масс-спектр гексадекана; 2 — 2-метнлпен-
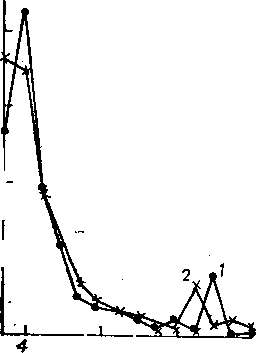
8 12 Число атомов С
тадекана
ионизации), перераспределяется ^ по вращательным, колебатель- § " ным и электронным состояниям. J &
Если в молекуле имеется сцсте- § : ма, благоприятствующая пере-\| даче возбуждения, например си- 8 _ стема сопряженных связей, то избыточная энергия успевает равномерно распределиться по всей молекуле и степень диссоциации подобных соединений оказывается сравнительно небольшой. При отсутствии подобной системы избыточная энергия не успевает перераспределиться по всему молекулярному иону, на одной из наиболее слабых связей в окрестности атома с локализованным положительным зарядом оказывается энергия, достаточная для разрыва, и происходит диссоциация.
Устойчивость молекул к электронному удару характеризуется относительным количеством нераспавшихся молекулярных ионов Wm'-
WK = Imoa/(!moa+ Yj /qck)’
где /мол н /ос* — количества молекулярных и осколочных ионов.
Структура образующихся ионов и их интенсивность находятся в качественной зависимости от строения молекул. Массы осколочных ионов, образующихся при диссоциативной иониза-' ции, можно предсказать на основании структуры молекул. И наоборот, по массам образующихся осколочных ионов можно судить о том, какие структурные элементы входили в состав исследуемого соединения.
Влияние структурных особенностей молекул анализируемых соединений на направления распада молекулярного иона может быть охарактеризовано кривыми интенсивностей ионов по числу углеродных атомов. На рис. 6.6 в качестве примера приведены кривые распределения для гексадекана (1) и 2-метилпентаде-кана (2). Кривая для гсксадекана имеет максимум, соответствующий ионам С4Н^ , и далее происходит плавное уменьшение интенсивностей пиков вплоть до молекулярного иона. Возникновение любого максимума на этой кривой означает наличие заместителя в молекуле. Так, при метальной группе в положении 2 на кривой распределения появляется максимум,, соответствующий ионам (М — СзН*)+; в частности, при диссоциации 2-метилпентадекана максимум отмечается для ионов
С13Н27, что объясняется меньшей энергией разрыва соответствующей связи С—С:
CH3(CH2)12-fiCHCH3 ,
¦ -X—1 I
. CjgH^ СН3т/е = 18b
Молекулярные ионы алканов неустойчивы, причем алканьг с разветвленной цепью еще менее устойчивы, чем н-алканы.. Например, для тетрадекана и 2-метилтридекана значения Wm равны 1,34 и 0,38%.
Циклоалканы несколько более устойчивы к электронному удару, чем алканы, причем шестичленные циклы стабильнее пятичленных, а бициклические алканы более стабильны, чем-моноциклические. Пятичленные циклоалканы образуют интенсивный пик с массовым числом (М — 28)+ и менее интенсивный пик (М — 70)+, соответствующий отщеплению радикала и миграции водорода:
Для шестичленных циклоалканов характерен пик (М — 83) +2 { <*•
Ароматические углеводороды легко ионизируются, так как имеют низкие потенциалы ионизации, но распад молекулярных, ионов идет сравнительно слабее. Так, для бензола U?m = 33%„ для хризена 48%. Наиболее вероятное направление распада' алкилбензолов— по p-связи, которое сопровождается и миграцией водорода:
L—QHs—СН3: (М-02)+
В масс-спектрах сложных смесей можно выделить групп» ионов (для алканов — пики ионов СпНгп+1 , для алкилбензолов— СпН2п-7 и т. п.), определяющиеся некоторыми структурными фрагментами молекул. Совокупность групп ионов, на которые разбивается исходный масс-спектр, можно изобразить в виде линейчатого спектра, положение линий которого соответствует положениям центров групп, а высота линий — суммарным интенсивностям пиков ионов каждой группы. Представление масс-спектров сложных смесей в виде групповых масс-спектров позволяет проводить с ними операции, как со спектрами индивидуальных соединений.
В бензиновых фракциях методом масс-спектрометрии определяют содержание н-алканов и изоалканов, циклопентановых и циклогексановых углеводородов, алкилбензолов. В керосино-газойлевых и масляных фракциях определяют алканы, моно-, би- и трицикланы, алкилбензолы, инданы и тетралины, алкил-нафталины, аценафтены и дифенилы, аценафтилены и флуо-рены, фенантрены и антрацены, бензотиофены. С помощью масс-спектрометрии можно оценивать такие структурные характеристики молекул, как степень конденсации колец, средняя длина заместителя, средняя степень замещения.
Алкены и циклоалканы образуют одинаковые характеристические пики, поэтому для их раздельного определения снимают масс-спектры двух образцов — исходного и после удаления алкенов обработкой серной кислотой.
Метод хромато-масс-спектрометрии — комбинирование газовой или жидкостной хроматографии, позволяющих разделять анализируемую фракцию на компоненты, с масс-спектрометрической идентификацией. Создание приборов типа Хромасс позволяет определять структуру индивидуальных компонентов нефти и их содержание.
6.5. УЛЬТРАФИОЛЕТОВАЯ И ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
Ультрафиолетовую и инфракрасную спектроскопию широко используют при анализе нефтей.
Поглощение энергии в ультрафиолетовой области обусловлено изменениями энергетического состояния внешних электронов. В органических соединениях такое поглощение связано с переходом валентных а- и я-электронов со связывающих орбиталей на соответствующие разрыхляющие, а также с переходами электронов неподеленных пар гетероатомов (п-электронов) типа п-+- п* и п —*¦ о*.
Последовательность энергетических уровней электронов следующая: разрыхляющая а*-орбиталь > разрыхляющая я*-орбиталь > несвязывающая л-орбиталь > связывающая л-ор-биталь > связывающая а-орбиталь.
Полосы поглощения в электронном спектре характеризуются длиной волны (А,) и интенсивностью поглощения. Интенсивность полос поглощения определяется вероятностью электрон-лого перехода, измеряется она обычно величиной молярного
Рис. 6.7. Кривые поглощения основных типов аренов в УФ-об* ласти:
1 — моноциклические; 2 — бициклические?. 3 — полициклические нелинейно конденсированные; 4 — полициклические линейно конденсированные
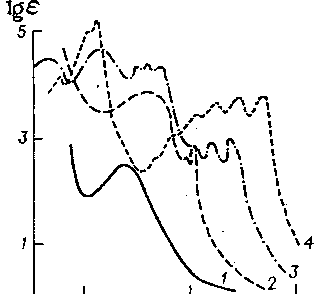
коэффициента поглощения в-максимуме полосы (еМакс или
1^Емакс)-
В молекулах насыщенных углеводородов возможны только переходы о->о*, требующие наибольшей энергии.
______ Полосы, соответствующие этим
240 320 400 переходам, лежат в дальней
Л,нм ультрафиолетовой области, поэтому для анализа содержания-насыщенных углеводородов требуется сложная аппаратура. Ал-кены и алкины с изолированными двойными связями имеют полосу поглощения также в области до 190 нм, обусловленную переходом я->я*. Для идентификации же компонентов нефтяных фракций используют спектры поглощения
в средней ультрафиолетовой области (К = 190—400 нм).
Сопряжение двойных связей вызывает смещение полос поглощения в длинноволновую сторону с одновременным увеличением их интенсивности. В средней УФ-области поглощают кг арены. Таким образом, УФ-спекгроскопию можно использовать, для анализа полиеновых и ароматических структур, остальные-углеводороды «прозрачны» в средней ультрафиолетовой области. При анализе продуктов термической переработки нефтяных фракций, в которых возможно присутствие полиенов, их_ необходимо предварительно отделить от ароматических углеводородов.
УФ-спектры аренов, как следует из рис. 6.7, существенно различаются в зависимости от числа циклов и линейного (типа-антрацена) или нелинейного (типа фенантрена) характера их конденсации. Максимум поглощения моноциклических аренов-находится в области 255—275 нм, для бициклических аренов-характерна более интенсивная полоса с максимумом 275— 290 нм и два близких пика в области 310—330 нм.
На основе усредненных спектральных данных по ароматическим ядрам разного типа получены уравнения для расчета-массового содержания бензольных С о, нафталиновых С„, фе-нантреновых Сф углеводородов, %:
- 0,255Кз75 + 0,022/^4351
Сф = — 0,001/Clea — 0,001/^230 "Н 0,391/^255 — 0,121/^270 "Н 0,023/^338 —
— 6.312/C37S + 0,710^435,
где К198, К2зо, ^255, Кш, Кззв, ^Сз75. ^435—удельные коэффициенты поглощения, л/(г-см), исследуемой фракции на аналитических длинах воли 198, 230 им и т. д. соответственно.
Предложены аналогичные уравнения и для расчета содержания в нефтяных фракциях антраценовых, пиреновых, хризе-новых (совместно с бензфлуореновыми) углеводородов, а также перилена. Приведенные выше уравнения позволяют рассчитывать содержание бензольных и полициклических углеводородов при их совместном присутствии.
В качестве растворителя при анализе аренов чаще всего применяют изооктан, очищенный на силикагеле.
Благодаря высокой чувствительности УФ-спектроскопия находит применение для определения следов аренов в неароматических продуктах. Наличие во фракции гетероатомных соединений сильно увеличивает поглощение в УФ-спектре и может привести к значительным погрешностям анализа.
В инфракрасной области, в .отличие от средней ультрафио-. летовой, поглощают все органические соединения. Эта область электромагнитного спектра связана с колебаниями атомов в молекулах. Каждая структурная группа характеризуется своим набором полос поглощения, число, положение и интенсивность которых в большей или меньшей степени зависят от состава остальной части молекулы. Для определения группового состава сложных смесей используют обычно характеристические, т. е. интенсивные полосы, при е > 10 моль/(л-см) практически сохраняющие интенсивность и общий вид независимо от строения остальной части молекулы; положение характеристических полос меняется в небольших пределах — до полуширины полосы.
ИК-спектры можно использовать для определения типа нефтей. Мерой содержания аренов служит площадь (Si) полосы v == 1610 см~\ обусловленной колебаниями связей С = С ароматического кольца, а мерой содержания алканов — площадь (S2) полосы v = 725 см-1, характеризующей колебание связей С—С в длинных цепях. Отношение A =Si/S2 принято за показатель ароматизированности нефтей. Нафтеновые структуры по ИК-спектрам не выявляются. Для метановых нефтей А < 0,35, метано-нафтеновых 0,3 ^ А ^ 0,5, нафтеновых 0,6 < А < 1,2, нафтено-ароматических 1,2 ^ А ^ 3,5.
Применение ИК-спектроскопии для структурно-групповогс анализа высококипящих (выше 200°С) алкано-циклоалкановых фракций позволяет получать количественные характеристики структурных фрагментов гипотетической средней молекулы. По характеристическим полосам поглощения в области 720— 780 см-1 рассчитывают среднее содержание метиленовых групп в алкильных цепях различной длины (этильных, пропильных радикалах и т. д.). По интегральным интенсивностям полос поглощения 1378 и 1366 см-1 можно приблизительно определить содержание изолированных и геминальных (т. е. находящихся при одном углеродном атоме) метильных групп. Однако точность этих определений невелика, так как в расчетах используют усредненные значения коэффициентов погашения для различных углеводородов. По полученным данным можно приблизительно оценить степень разветвленности алифатических цепей.
С использованием коэффициентов погашения исследуемой фракции на аналитических частотах 2926 и 2957 см-1 рассчитывают содержание метиленовых групп в пяти- и шестичленных насыщенных кольцах.
Достаточно широко используют ИК-спектроскопию и - для исследования гетероатомных соединений нефти после ее выделения и разделения на узкие фракции. В ИК-спектрах сырых нефтей и их фракций обнаруживаются практически все характеристические полосы поглощения основных функциональных групп. Многокомпонентность состава, внутри- и межмолекуляр-ная структура нефтяных систем обусловливают сложную картину перекрывания и наложения полос поглощения с искажением их формы и интенсивности. Поэтому прямая идентификация и тем более количественное определение функциональных групп но интенсивностям поглощения в ИК-спектрах оказываются невозможными. Однако возможности ИК-спектроскопии расширяются по мере развития методов разделения нефти на однотипные группы компонентов.
6.6. ЯДЕРНЫЙ МАГНИТНЫЙ
И ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС
Метод ЯМР широко применяется для исследования структуры органических соединений наряду с методами оптической спектроскопии. Поглощение энергии радиочастотного излучения, которое используется в этом методе, связано с магнитными свойствами ядер.
Для получения спектров ЯМР образец помещают в сильное однородное магнитное поле и действуют на него радиочастотным излучением. Изменяя частоту генератора, возбуждающего магнитое поле, перпендикулярное к постоянному полю магнита, достигают условия резонансного поглощения энергии. Резонанс-
ная частота зависит от напряженности постоянного магнитного поля и значения магнитного момента ядер. Наиболее широко в исследованиях органических соединений, в том числе нефти, применяется протонный магнитный резонанс (ПМР).
Спектры ПМР характеризуются значениями химических сдвигов протона. Химическим сдвигом называется расстояние между резонансными сигналами протонов образца и стандарта— тетраметилсилана. Это расстояние зависит от напряженности магнитного поля (или частоты), поэтому химический сдвиг измеряется в относительных единицах — миллионных долях (м. д.) поля или резонансной частоты. Химический сдвиг зависит от структуры молекул — электронной плотности у протона и напряженности вторичных магнитных полей,' возникающих вследствие движения электронов соседних атомов.
По мере усовершенствования техники метода ЯМР и увеличения рабочей частоты спектрометров до 100—220 МГц повышается селективность определения протонов в различных структурах. Метод ПМР дает информацию о распределении водорода, связанного с ароматическими циклами, гетероатомами, а также входящего в состав метальных, метиленовых и метановых групп. Особый интерес представляет применение метода ЯМР для исследования высококипящих нефтяных фракций.
Типичный спектр 'Н ЯМР нефтяной фракции (рис. 6.8) может быть разделен на четыре области. Область А(6,5—8,5м.д.) соответствует сигналам ароматических протонов; область ор (1,8—4,0 м. д.)—протонов СН-, СН2-, СНз-групп, находящихся в a-положении к ароматическим ядрам; область Р (1,0— 1,8 м. д.)—метиленовых и метиновых протонов, удаленных от ароматических ядер, а также групп СНз в P-положении к ароматическим ядрам; область у (0,7—1,0 м. д.) — протонов метальных групп, более удаленных от ароматических ядер. Пло-
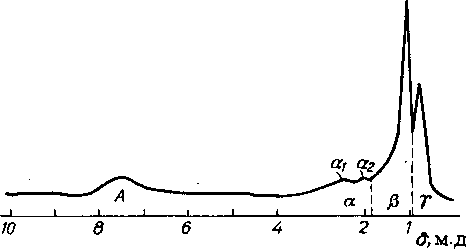
Рис. 6.8. Типичный спектр ‘Н ЯМР нефтяной фракции
Обозначения см. в тексте
щади областей пропорциональны количеству протонов, дающих эти сигналы.
Если известна средняя эмпирическая формула, рассчитываемая по элементному составу и средней молекулярной массе фракции, то можно распределить атомы водорода по структурным группам. Спектроскопия 'Н ЯМР позволяет получать большое количество структурно-групповнх характеристик «средней молекулы». Недостаток метода состоит в том, что особенности строения углеродных скелетов приходится рассчитывать по распределению водорода, вводя ряд допущений и приближений.
Единственный метод, позволяющий непосредственно измерить долю ароматического углерода — 13С ЯМР-спектроско-ция. Типичный спектр 13С ЯМР нефтяной фракции (рис. 6.9) содержит широкие полосы поглощения атомов углерода в насыщенных (0—70 м. д.) и ароматических (100—170 м. д.) структурах. Проинтегрировав спектр, рассчитывают фактор ароматичности:
fa — + Лас)>
где /а > /нас — интегральные интенсивности гиков, относящихся к ароматическим и насыщенным структурам соответствешо.
По отношению интегральных интенсивностей пиков частот 29,7 и 14,1 м. д. можно рассчитать длину цепи алкильного заместителя:
п = hgj/Iuti -f- 7.
Аналитические возможности метода ЯМР постоянно ув;ели-чиваются благодаря совершенствованию спектрометров и разработке новых методов получения спектров ЯМР.
Электронный парамагнитный резонанс (ЭПР) открыт в 1944 г. Е. К. Завойским. Парамагнетизмом обладают системы, на электронных оболочках которых имеются неспаренные электроны. К таким системам относятся свободные радикалы, парамагнитные ионы, внедренные в кристаллическую решетку или в молекулы комплексных соединений, и т. д. Как
29,7
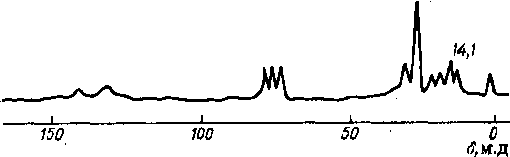
Рис. 6.9. Типичный спектр ,ЭС ЯМР нефтяной фракции 144
оказалось, парамагнетизмом обладают и нефти, благодаря входящим в них смо-листо-асфальтеновым компонентам.
ЭПР-спектр снимают, воздействуя на образец, помещенный в сильное магнитное поле, сверхвысокочастотным полем генератора. ЭПР-спектр (рис. 6.10) представляет собой одиночный сигнал, интерпретация которого сводится к расчету числа парамагнитных центров Nx в образце путем сравнения производной кривой поглощения образца и эталона. Эталон— сахарный уголь — имеет стабильное значение числа парамагнитных центров (Л/эт = 0,1 • 10‘7 на 1 г). Число
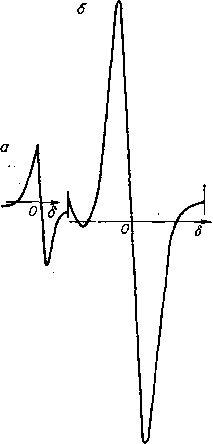
парамагнитных центров образца рассчитывают по формуле:
Нх = Мэт1хИН2х!(1этИН2вгт),
где 1Х — амплитуда производной сигнала поглощения образца; ДЯ* — ширина производной сигнала поглощения образца; т — масса образца.
ЭПР-спектры нефтей позволяют провести сопоставительный анализ степени их обогащенности смолисто-асфальтеновыми компонентами. Ширина ЭПР-сигнала отражает степень уплотнения структуры асфальтенов: чем она выше; тем меньше ширина сигнала.
