Использование заводских углеводородных газов. заводы топливного и комплексного профиля глава viii использование и переработка заводских углеводородных газов характеристика газов
ИСПОЛЬЗОВАНИЕ ЗАВОДСКИХ УГЛЕВОДОРОДНЫХ ГАЗОВ. ЗАВОДЫ ТОПЛИВНОГО И КОМПЛЕКСНОГО ПРОФИЛЯ
ГЛАВА VIII
ИСПОЛЬЗОВАНИЕ И ПЕРЕРАБОТКА ЗАВОДСКИХ УГЛЕВОДОРОДНЫХ ГАЗОВ
ХАРАКТЕРИСТИКА ГАЗОВ
Все процессы деструктивной переработки нефтяного сырья сопровождаются образованием углеводородных газов. Выход этих газов составляет в среднем 5—20% на сырье. При глубокой переработке современный нефтеперерабатывающий завод мощностью 12 млн. т нефти в год дает примерно 1 млн. т (т. е. свыше 8% масс.), газообразных углеводородов. Особое место среди деструктивных процессов занимает в этом отношении пиролиз, где газ, богатый легкими олефинами, является целевым продуктом. В этом случае после извлечения этилена, пропилена и бутилен-бутадиено-вой фракции также остается насыщенная часть газа, которая при пиролизе газов в основном идет на рециркуляцию, а при пиролизе бензина и другого жидкого сырья уходит с газофракционирующей установки.
Выходы газа при основных каталитических процессах перера-¦ ботки нефтяного сырья весьма значительны: каталитический риформинг дает 10—20% (масс.) газа на сырье (в том числе от 1 до 2% водорода); при каталитическом крекинге выход газа составляет 12—15% (масс.). В табл. 39 дан примерный состав газов, образующихся при основных процессах нефтепереработки.
Для процессов, протекающих под давлением водорода (риформинг, изомеризация, гидрокрекинг, гидроочистка), состав газов относительно несложен и подобно природным и попутным газам характеризуется отсутствием непредельных углеводородов. В то же время все термические и часть каталитических процессов дают газы более сложного состава, с большим или меньшим содержа-, яием непредельных углеводородов. Концентрация непредельных углеводородов в некоторой степени зависит от состава сырья, но главным образом определяется жесткостью режима, а для каталитического крекинга — и применяемым катализатором. Например, непрерывное коксование гудрона при обычном режиме (530—
540 °С) дает газ с «30% (масс.) непредельных углеводородов, а повышение температуры до 600 °С увеличивает сумму непредельных почти до 50%. Переход установок каталитического крекинга на цеолитсодержащие катализаторы вызвал снижение общего выхода газа.
Помимо содержания непредельных углеводородов заводские газы характеризуются еще концентрацией «жирной» части — фракции С3—С4. Наиболее ценные углеводороды этой фракции — изо-бутан и бутилены, являющиеся сырьем каталитического алкили-рования (получение высокооктанового компонента автомобильных и авиационных бензинов). Наименьший интерес представляет «сухая» часть газа — водород, метан и фракция Сг (этан+этилен). Входящие в состав сухого газа водород и этилен представляют ценность, но водород извлекают только из газа риформинга, так как он образуется там в значительных количествах и отделяется в газосепараторе высокого давления на самой установке риформинга (остальной газ содержит только следы водорода). Газы остальных процессов при их смешении до очистки и газофракцио-нирования содержат водород уже в относительно небольшой кон-
Таблица 39. Состав углеводородных газов при основных процессах нефтепереработки
Состав газа, % (масс.)
|
Компоненты | Термический крекинг под давлениема | Замедленное коксование |
непрерывное коксование в псевдоожиженном слое кокса |
каталитический крекинг вакуумного газойляг |
каталитический риформинг бензниа^ |
гидрокрекинг тяжелого 1 | дистиллятного сырьяе j | ||
| гудрона® | л® » ? я р й « 0J г. & § | на аморфном катализаторе РСГ-2 | на цеолит-содержащем катализаторе РСГ-2Ц | |||||
| Водород |
0,2 | 0,4 |
0,6 | 0,6 |
3,2 | 0,1 |
6,0 | |
|
Метан | 16,0 |
35,9 | 42,5 |
23,2 | 8,3 |
3,4 | 13,0 |
6,9 |
| Этилен | 2.5 | 1,7 |
1,5 | 18,3 |
7,1 | 4,5 | — | — |
| Этан | 17,0 | 18,2 |
20,1 | 15,3 |
2,9 | 2,8 |
21,0 | 14,0 |
| Пропилен | 9,0 |
5,9 | 0,9 |
17,4 | 26,6 |
23,8 | — | — |
| Пропан |
21,5 | 17,0 |
17,9 | 9,2 |
5,9 | 10,7 |
32,0 | 44,7 |
| н-Бутилен | 9,8 | 3,7 | 1,3 |
7,7 | 16,6 |
15,9 | — | — |
| 'н-Бутан |
14,5 | 9,3 |
11,0 | 2,5 |
3,1 | 5,8 |
16,0 | 10,4 |
| Изобутан | 5,0 |
5,6 | 3,4 |
0,6 | 15, $ |
25,2 | 12,0 |
24,0 |
|
Изобутилен | 4,5 |
2,3 | 0,8 |
5,2 | 10,5 |
7,8 | ' ’ |
- |
| Сумма непредельных | 25,8 |
13,6 | 4,5 |
48,6 | 60,8 |
52,0 | — | — |
а Данные А. Н. Тарасова. ^Данные А. Ф. Красюкова. в Данные 3. И. Сюняева. г Данные В. Н. Еркииа.д Обобщенные данные. е Данные С. П. Рогова.
центрации. При глубокой переработке нефти выход сухих газов достигает 3—4,5% (масс.), а состав их примерно следующий (%. масс, на газ):
Водород.....3,0—3,5 Этан......«30
Метан..........26—27 Пропан+пропилен . . 8,0—8,5
Этилен..........27—28 Фракция С* . . . . «5
Разумеется, состав сухого газа на разных заводах колеблется в зависимости от профиля завода и соотношения между мощностями отдельных процессов. Сложность технологии выделения этилена и водорода заставляет пока отказаться от этого, и сухой газ используют обычно на заводе в качестве технологического топлива. Однако не исключено, что будет предусмотрено предвари-тельное выделение наиболее ценных компонентов из сухого газа. М, По относительной концентрации сухой и жирной части газа можно считать «сухими» газ термического крекинга под давлением Н и газ коксования, где содержание фракций до Сг включительно В составляет 35—60% (масс.). Напротив, газы каталитического кре- Н кинга содержат 60—75% углеводородов Сз—С4 (см. табл. 39) — ™ это «жирные» газы.
Ресурсы нефтезаводских газов, естественно, связаны с глубиной переработки нефти на заводе; при глубокой переработке рациональное использование газа имеет особенно большое экономическое значение. Направление переработки газовых фракций л определяется профилем завода, особенно с учетом того, что завод Я обычно представляет собой нефтехимический комплекс, в котором Я процессы нефтепереработки сочетаются с подготовкой мономеров Я для нефтехимического синтеза или сопровождаются самими нефте- В химическими процессами (получение полипропилена, присадок Я
Некоторые газовые компоненты используют непосредственно уВ на заводе: «сухой» газ обычно является технологическим топли Я вом, водородсодержащий газ риформинга необходим для гидрон В низационных процессов (гидроочистка, гидрокрекинг). Если в схе Я му завода включены установки гидрокрекинга, потребность в водо- Я роде не может быть удовлетворена одним риформингом, поэтому Я часть сухого газа (обычно метан и этан) подвергают конверсии В с целью получения водорода. И
Существенным для использования нефтезаводских газов ;Я является полнота отбора наиболее ценных компонентов от их по- Вт тенциального содержания, т. е. эффективная работа установок В ¦газоразделения. На большинстве современных НПЗ имеется два В блока газоразделения: для предельных и для непредельных газов. Ш Совместное разделение этих газов нерационально, так как непре- Я дельные компоненты более ценны, и их легче отобрать с наиболь- Я шей полнотой из более концентрированных смесей. Схемы газо- В
Таблица 40. Некоторые физические константы газообразных углеводородов
| Углеводород |
Т. кип. при 760 мм рт. ст., °С |
ПЛОТНОСТЬ газа. КГ/мЗ |
Плотность сжиженного газа,* кг/мЗ |
Критическая температура, °С | Критиче ское давление, МПа |
|
Метан | -161,6 | 0,7166 | 380 | —82,1 |
4,58 |
| Этилен |
—103,7 | 1,2594 | 569,9 (—103,4 °С) | 9,5 | 5,07 |
| Этан | —88,6 | 1,3561 | 374 |
32,3 | 4,82 |
|
Пропилен | —47,7 |
1,8753 | 609,5 (—47 °С) |
91,4 | 4,54 |
|
Пропая | —42,1 |
2,0193 | 500,7 | 96,8 |
.4,20 |
| Изобутилен | —7,0 | 2,5001 |
626,8 (—6,8 °С) | 144,7 |
3,95 |
| а-Бутилен |
—6,3 | 2,5001 | 625 (-6,1 °С) | 147,2 | 4,00 |
| зуигяс-.р-Бутилен |
0,9 | — | 604,2 | — | — |
| цыс-|3-Бутилен | 3,7 |
— | 621,3 | — | — |
| Изобутан | — 11,7 | 2,6720 |
557,2 | 134,4 | 3,64 |
| я-Бутан | —0,50 | 2 6720 | 578,7 |
152,0 | 3,75 |
|
Ацетилен | —83,6 |
1,1774 | 615,4 (—80,3 °С) |
35,3 | _ 6,11 |
|
Бутадиен | -4,6 |
2,4353 | 620,6 | 163,2 |
* В скобках указана температура, при которой определена плотность сжиженного
газа.
разделения предельных и непредельных газов могут быть аналогичными или несколько различаться (в зависимости от профиля завода, т. е. от удельного объема тех и других газов).
В табл. 40 представлены некоторые физические константы газообразных углеводородов. Наименее летучими являются изомеры {5-бутилена и н-бутан. Критические температуры компонентов фракции С4 лежат в пределах 134—163 °С, что свидетельствует о возможности ожижения этих углеводородов при относительно низких давлениях и температурах выше 30—40 °С, доступных для водяного охлаждения. Например, н-бутан при 40 °С имеет давление пдров «0,4 МПа, и при этой температуре может быть легко превращен в жидкость (даже при наличии водяного охлаждения вверху бутановой колонны). Наоборот, этилен имеет критическую температуру всего +9,5 °С, т. е. для его конденсации даже при значительно более высоком давлении водяное охлаждение непригодно, и требуется применять специальные хладоагенты.
ПОДГОТОВКА ГАЗОВ К ПЕРЕРАБОТКЕ
Общие принципы и технологические схемы осушки углеводородных газов и очистки их от сероводорода изложены в части I курса «Технология переработки нефти и газа» применительно к природным и попутным газам. Ниже упомянуты только те методы подготовки, которые свойственны заводским углеводородным газам.
Осушка заводских газов требуется не всегда. Как правВило е применяют в тех случаях, когда газ подвергают последующей низ котемпературной ректификации (например, при выделении i чистог этилена) или направляют непосредственно для каталии-ическод переработки на установку с катализатором, чувствительным^ к влаге. При низких температурах ректификации (до —100 °С) водный конденсат будет выпадать даже при небольшой влажности газа Например, для углеводородного газа, находящегося при 0,МПа' при содержании воды 2 г/м3 точка росы была л; 14 °С, а 1ПрИ С0Г держании воды 0,17 г/м3 всего-—20°С, т. е. при темперраТурах ниже —20 °С газ должен был содержать менее 0,17 г вл1аги на
1 м3. Повышение давления также вызывает необходимости более глубокой осушки, так как давление увеличивает точку рос;ы Например, для того же газа при увеличении абсолютного давления с 0,7 до 3,5 МПа точка росы повысилась с 14 до 39 °С, а пр^и 14 °с и 3,5 МПа максимально допустимое влагосодержание составило всего 0,5 г/м3.
Степень осушки газа определяется не только возможностью конденсации воды, но и образованием гидратов. Гидраты! представляют собой комплексные соединения молекул газа с водой, Известны гидраты метана (СН4-6Н20), этана (С2Нб-7Н20)) и Др* По внешнему виду гидраты — объемистые кристаллические; образования, в зависимости от состава белые или прозрачные, к^к лед Гидраты нестабильны и при изменении температуры или даьпрния легко разлагаются на газ и воду.
Характерно, что гидраты способны образовываться только При повышенных давлениях и при температурах выше нуля, Причем более тяжелые углеводороды образуют гидраты легче, чем низкомолекулярные. Так, метан способен образовывать гидрагт при 12,5 °С и 10 МПа; этан при этой же температуре образует 1ГИДрат под давлением всего 2,5 МПа. Гидраты могут существовать Только при наличии избыточной влаги в газе, т. е. когда парциально>е давление паров воды в газовой фазе больше давления паров ги,Драта Таким образом, содержание влаги в газе должно соответст^овать" такой точке росы, при которой давление насыщенного во^яного пара будет меньше давления паров гидрата при темперзаТуре среды.
Жидкие поглотители влаги не могут снизить точку росы ниже минус 15 °С, а для надежной работы установки, фракционирующей газы пиролиза, точка росы не должна превышать мину^ gg_
минус 70 °С. Поэтому для осушки газов пиролиза испол ьзуют твердые поглотители — в основном цеолиты или цеолиты с а.дюмо-гелем. Осушка адсорбентами газов, содержащих непредеЛЬНЬ1е углеводороды, осложняется возможностью частичной полимьпИза. ции этих компонентов. Применительно к газу пиролиза исключительно большое значение имеет предварительное отделение углеводородов С5 и С4, состоящих частично из диенов, которые наиболее легко полимеризуются. Содержание в газе 3—5% (масс.) углеводородов С5 приводит к быстрой потере активности адсорбента.
Необходимым узлом установок каталитического риформинга является блок осушки циркулирующего водородсодержащего газа. В гл. VI отмечалось, что во избежание дезактивирования катализатора (за счет вымывания галогена) содержание влаги в циркулирующем газе поддерживают в пределах (14-1,5) 10~3% (об.).
Предварительную осушку сырья риформинга осуществляют в стабилизационной колонне блока гидроочистки. Осушка циркулирующего газа происходит в одном из двух или более поочередно работающих адсорберов, заполненных кислотоустойчивыми (против НС1) цеолитами. Время непрерывной работы адсорбера составляет 24—36 ч, после чего подачу газа переключают на другой адсорбер, а слой отработанного цеолита обрабатывают горячим инертным газом (до 350 °С). По данным А. Д. Сулимова, адсорберы для осушки газа могут быть включены только на период вывода установки риформинга на режим, а также при регенерации катализатора.
Как правило, прежде чем направить заводские газы на разде-_ ление, их подвергают очистке. Целью очистки чаще всего является удаление сернистых соединений, представленных в нефтяных газах в основном сероводородом. Присутствие сероводорода в газе недопустимо вследствие: 1) корродирующих и токсичных свойств сероводорода и 2) отравляющего действия на многие катализаторы. Поскольку при переработке сернистого сырья концентрация сероводорода в газе может быть весьма значительна, необходимо не только удалять его из газа, но и использовать для получения серы или серной кислоты. Если тяжелые газовые компоненты получают с технологической установки в жидком виде (под давлением), их иногда подвергают только промывке щелочью для удаления сернистых и кислотных соединений. Для очистки углеводородов, находящихся в газовой фазе, используют водные растворы этанолами-нов, фенолятов и других реагентов. Наиболее распространена очистка этаноламинами:
H2N-C2H4OH HN(C2H4OH)2 N(C2H4OH)3
моноэтаиоламнн диэтаноламнн триэтаноламин
Очистка газа этаноламинами является типичным примером кругового сорбционного процесса (рис. 101). В процессах такого типа сероводород поглощается из газа раствором реагента в одном аппарате и выделяется из раствора в результате его отпаривания в другом аппарате. Регенерированный таким образом реагент возвращают на поглощение сероводорода. Очистка газа происходит путем хемосорбции сероводорода 15—30%-ным водным раствором этаноламина. Иногда используют смесь этаноламина с эти^ленгли колем или осуществляют последовательную обработку газаа этим растворителями. Например, на установках каталитического } рифор минга более старого типа (35-5), где гидроочистка сырья о'ЭТСуТСТ вовала, была предусмотрена очистка циркулирующего газаа моно этаноламином с последующей промывкой водой и осушкой ДИЭти-
ленгликолем. Т^хно^ЛОгиче Очищенный CepoSodopol кая схема очистки заводски
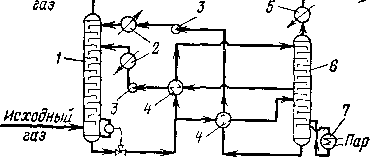
Рис. 101. Технологическая схема очистки газа с двухступенчатой подачей моноэтаноламина:
1 — абсорбер; 2— холодильники абсорбента; 3 — насосы; 4 — теплообменники; 5 — холодильник сероводорода; 6 — десорбер; 7 — кнпятнльник.
газов в принципе не о0тлича ется от таковой дляя природного газа.
Из других аминов i применяют метилдиэтанола^мин ^ N-метилпирролидон ({циклический амин); пос^Л0диий рекомендуется для ras30Bj содержащих помимо ^сероводорода значительное количество диоксида угллерода.
Основными аппаргатами для очистки газов жи^д^ими реагентами являются абсорбер тарельчатого или насадочного типа и отпарная колоннга (де-сорбер). Абсорбер изготавливают из углеродистой стали; в нем имеется 10—20 тарелок или насадка из колец Рашига. Отггонные колонны для отпаривания сероводорода также изготавлива!ют та. рельчатыми или насадочными.
Тепло, необходимое для отпаривания, вводят через выносной кипятильник, обогреваемый обычно водяным паром. Важнцщ параметром является температура низа десорбера. Так, для моноэтаноламина рекомендуется температура отпаривания не более 125 °С, поскольку при повышении температуры скорость раз,ложе_ ния этого реагента быстро возрастает. Вследствие относитгельно высокой температуры в низу отпарной колонны и в кипятил1ЬНИке наблюдается сероводородная коррозия, поэтому трубки кип%ТИЛь-ника изготавливают из нержавеющей стали (Ст. 18-8); н%жняя часть колонны также имеет соответствующую облицовку.
РАЗДЕЛЕНИЕ ГАЗА НА КОМПОНЕНТЫ
Состав заводских газов, представленный в табл. 39 (стр. 275), является типичным для всего газа (балансового количества) F получаемого при данном процессе, т. е. имеется в виду, что} getI. зиновая фракция при этом не содержит газообразных компонентов — бензин стабилен. Практически отделение газа от бе!НЗина можно осуществлять в одну или несколько ступеней.
Так, например, при каталитическом риформинге324 и при гидрогенизационных процессах в газосепараторе высокого давления отделяется водородсодержащий газ. Концентрация водорода в нем определяется давлением и температурой сепарации: чем выше давление и чем ниже температура, тем больше растворимость углеводородной части газа в катализате и тем «суше» (легче) отделяемый от катализата газ.
В газосепараторе высокого давления, размещенном по схеме установки каталитического риформинга непосредственно за теплообменниками и конденсаторами, давление почти такое же, как в реакторном блоке, например 3 МПа; это обеспечивает сепарацию газа с 70—80% (об.) водорода, а большая часть углеводородных компонентов остается растворенной в катализате. Составы газовой и жидкой фаз будут определяться равновесными соотношениями, присущими данному сочетанию температуры и давления. В следующем газосепараторе за счет перепада давления из катализата выделяется часть углеводородного газа, но наиболее тяжелая его часть остается (преимущественно) в катализате, который необходимо подвергать стабилизации. Аналогичная картина наблюдается и на гидрогенизационных установках.
В процессе каталитического крекинга, который идет при давлении, близком к атмосферному, низкое давление в газосепараторе заставляет прибегать к компрессору, на прием которого поступает газ. Однако и в этом случае, хотя режим газосепаратора благоприятствует отделению тяжелых компонентов, последние будут частично оставаться в бензине, и потребуется его стабилизация. В то же время газ, уходящий из газосепаратора, захватывает а легкие фракции бензина, которые должны быть затем из него извлечены.
Многие из современных схем сочетают разделение газа на компоненты и стабилизацию бензина.
Для четкого разделения газообразных углеводородов требуется ректификация или сочетание ректификации с абсорбцией. Последнее необходимо, если в газе много «сухой» части, особенно метана. В этом случае целесообразно вначале отделить «сухую» часть посредством абсорбции с последующим разделением остального газа ректификацией. При умеренном содержании метана узел абсорбции можно из схемы исключить. На рис. 102 представлена схема такого типа, предназначенная для разделения предельных газов: жирного газа и нестабильной головной фракции с установок атмосферной перегонки нефти, а также газов каталитического риформинга.
Все газообразные компоненты (углеводородный газ риформинга, газ прямой перегонки) подвергают компримироганию компрессором ЦК-1) с последующим охлаждением в водяном холодильни-нике ХК-1 и аммиачном холодильнике ХК-2 до 4ЧС. К сжиженному газу в сборнике С-4 присоединяют нестабильные жидкие отгоны стабилизации с установок АТ и риформинга и направляют весь поток в колонну-деэтанизатор К-1-
Колонна К-1 работает в режиме неполной конденсации головного продукта — метан-этановой фракции. Аммиачный холодиль-
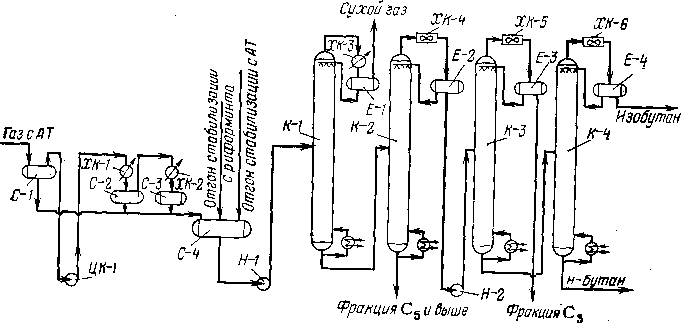
Рис. 102. Технологическая схема газофракционирующей установки (ГФУ):
ЦК-1 — газовый компрессор; ХК-1 — водяной холодильник; ХК-2, ХК-3 — аммиачные холодильники; ХК-4, ХК-5, ХК-6 — воздушные холодильники; С-1, С-2, С-3, С-4 — сепараторы-сборники жидкого газа; К-1 — деэтанизатор; К'2 — дебутанизатор; К-3 — пропановая колонна; К-4 — изобутановая колонна; Е-1, Е-2, Е-3, Е-4 — емкости орошения; Н-1, Н-2 — насосы.
ник ХК-3 охлаждает головной погон до 0°С; при этом' конденсат (более тяжелая часть сухого газа — этан) циркулирует в виде орошения, а балансовое количество сухого газа уходит с верха емкости Е-1. Деэтанизированный остаток из колонны К-1 поступает на дальнейшее разделение в колонну К-2. Колонна К-2 служит для отделения пропан-бутановой фракции от углеводородов Cs и выше. Головной погон колонны К-2 после конденсации и охлаждения частично служит орошением этой колонны; остальной конденсат поступает в пропановую колонну К-3, где отделяется пропановая фракция. В колонне К-4 происходит разделение н- и изо: бутана.
Ниже представлены основные режимные показатели колонн описанной ГФУ и число тарелок в колоннах:
| Колонна |
Температура, °С | Давление. | Число | |
|
верх | низ | МПа | тарелок | |
|
К-1 | 26—30 |
130—140 | 2,6 | ' 45 |
| К-2 | 70 | 150—160 | 1,3 |
45 |
| к-з |
60 | 114 | 2,2 |
60 |
| К-4 |
68 | 83 | 1,1 |
60 |
Установки фракционирования газов путем ректификации ха* рактеризуются некоторыми особенностями. Необходимость полной или частичной конденсации головного погона заставляет осуществлять ректификацию под давлением, которое тем выше, чем легче головной погон. Однако повышенное давление затрудняет разделение. Например, для бинарной смеси пропан+изобутан относительная летучесть а при 100 ЧС и 2 МПа равна «1,7, а при той же температуре, но при 1 МПа уже а=1,9, т. е. разделение облегчается.
Последующее использование компонентов газа требует достаточно четкого их разделения и высокого отбора от потенциала, поэтому колонны ГФУ содержат большое число тарелок. Известно, что допустимая скорость паров в колоннах является функцией разности плотностей горячей флегмы, стекающей с тарелки, и паров, поднимающихся в том же сечении. Поскольку повышение давления до 1—2 МПа увеличивает плотность паров соответственно в 10—20 раз (против условий разделения при атмосферном давлении), допустимые скорости паров в колоннах ГФУ не превышают 0,20—0,25 м/с.
Описанная схема ГФУ мало пригодна, если газ богат метаном, что свойственно, например, газам термического крекинга и коксования. В этом случае в емкости орошения первой колонны (деэта-низатор) вследствие высокого парциального давления метана не удается достигнуть даже частичной конденсации газа. Колонна работает только как испаритель, и в схему газофракционирования необходимо включить узел предварительного абсорбционного выделения метан-этановой фракции, т. е. разделять газ по абсорбционно-ректификационной схеме (АГФУ).
Применение обычной абсорбции недостаточно эффективно, так как только абсорбцией нельзя добиться четкого разделения, и если сухой газ будет отобран на 100% от потенциального содержания в газовой смеси, он неизбежно захватит с собой некоторое количество более тяжелых компонентов. Если же пойти на то, чтобы в сухом газе совершенно отсутствовала фракция Сз, часть сухого газа уйдет вместе с насыщенным абсорбентом и при отпаривании последнего попадет во фракции Сз—С4.
Наиболее широкое применение нашел в настоящее время аппарат, получивший название фракционирующего абсорбера и сочетающий процессы абсорбции фракции Сз и десорбции сухого газа (поэтому фракционирующий абсорбер называют иногда абсор-бером-десорбером). Фракционирующий абсорбер представляет собой комбинированную колонну; в верхнюю ее часть поступает холодный абсорбент, а в нижней сообщается тепло. Жирный газ подают в среднюю часть аппарата (рис. 103). Обычно в аппарате имеется 40—50 тарелок, распределенных примерно поровну между абсорбционной и десорбционной секциями. В результате много-
ступенчатого контакта газовой и жидкой фаз в верхней ч части аппарата поглощается наиболее тяжелая часть газа; стекая е вниз, насыщенный абсорбент встречается со все более горячими парфами, десорбированными из жидкости, стекающей в нижнюю час’тыъ колонны. В результате с верха фракционирующего абсорбера ухосодит
сухой газ, содержащий углеводорроды Ci—С2, а снизу вместе с тощим а&бсор-бентом выводят углеводороды С3-з—С4.
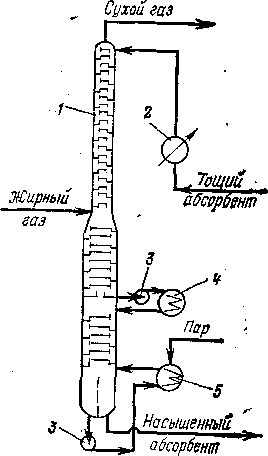
Рис. 103. Фракционирующий абсорбер (деэтанизатор):
I —колонна; 2, 4 — холодильники «бсорбента; 3— насосы; 5 — кипя
тильник.
Давление во фракционирующекм абсорбере поддерживают обычно отэт 1,2 до 2,0 МПа, хотя в некоторых слуучаях оно достигает 3 МПа. При повышлении давления поглощение газовых шомпо-нентов возрастает, но следует имаеть в виду, что повышение давления в прреде-лах 1,2—2 МПа мало способствуем поглощению пропана, и в то же врремя значительно увеличивается недакела-тельная абсорбция этана (констганты равновесия углеводородов Си—Сг уменьшаются с ростом давленияя в большей степени, чем для углеводдоро-дов Сз—С4).
Ниже описана схема устаноовки* совместного газоразделения и сггаби-лизации бензина каталитического) крекинга, эксплуатируемой на однош из заводов (рис. 104). Основными гаппа-ратами являются газофракционкирую-щий абсорбер 3, стабилизационная колонна 8, пропановая колонна 11 ш бу-тановая колонна 14.
Жирный газ из газосепараторга через верх каплеотбойника / поступает на блок очистки А монюэта-ноламином и потом компрессорами подается в газофракциоширу-ющий абсорбер 3; туда же в качестве орошения подают насосом нестабильный бензин с низа емкости 2, а также (несколько выше ввода газа) конденсат, образовавшийся в результате компрессии жирного газа, и жидкость из каплеотбойника /.
Основным абсорбентом, подаваемым на верх абсорбера 3, служит нестабильный бензин из емкости 2\ дополнительно для поглощения уноса нестабильного бензина подают стабильный бензин (несколькими тарелками выше). В абсорбере имеется система
трех циркуляционных орошений для съема тепла абсорбции; циркулирующие потоки охлаждают в водяных холодильниках 4 и возвращают на вышележащую тарелку. Сухой газ проходит через газосепаратор 5, где отделяется некоторое количество конденсата» и уходит в газовую сеть завода.
Сухой газ
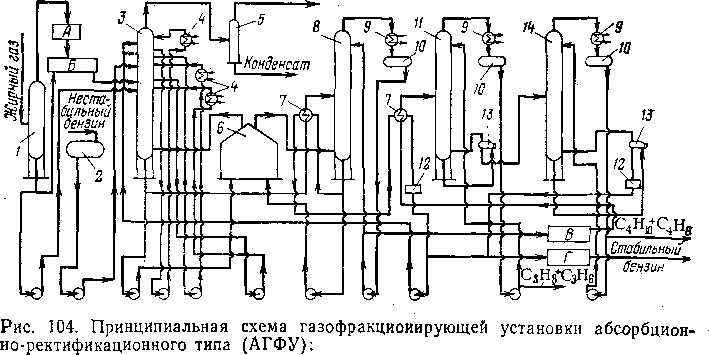
1 — каплеотбойник; 2, 10— емкости; 3 — газофракционирующий абсорбер; 4 — холодильники циркуляционного орошения; 5 — газосепаратор; 6 — трубчатая печь (рибойлер); 7 — теплообменники; 8 — стабилизатор; 9 — холодильники-конденсаторы; 11 — пропановая колонна; /2—. холодильники; 13 — рибойлеры; 14 — бутановая колонна; А — блок очистки газа моноэтанол» амином; Б — компрессорная; В — блок очистки и осушки отгона стабилизации; Г — блок защелачивания стабильного бензина.
Деэтанизированный бензин с поглощенными фракциями С3—С4 подогревают в теплообменнике 7 и подают в стабилизационную колонну 8, назначением которой является дебутанизация бензина. Печь 6 (двухсекционная) является рибойлером для колонн 3 к 8. Стабильный бензин проходит через теплообменник 7, отдает тепло нестабильному бензину и сырью пропановой колонны, охлаждается в холодильнике 12 и направляется в блок Г защелачивания. Отгон (головка) стабилизации конденсируется в холодильнике-конденсаторе 9 и из емкости 10 частично откачивается на орошение колонны 8\ балансовое количество отгона направляют последовательно на очистку моноэтаноламином и раствором щелочи и на осушку диэтиленгликолем. Затем отгон, состоящий в основном из фракций Сз—С4, направляют в колонну И для отделения пропан-пропиленовой фракции, которая с верха этой колонны после конденсации и охлаждения выводится с установки.
Остаток из колонны 11 перетекает в колонну 14, где происходит аналогичное отделение бутан-бутиленовой фракции от более тяжелого остатка (в основном фракция С5), который через холодильник 12 присоединяется к потоку стабильного бензина. Ввиду
Таблица 41. Характеристика аппаратов и технологический режим газофракционирующей установки
Абсорбер 3 Колонна 8 Колонна J1 Колонна 14
Показатели
Диаметр, мм
Расстояние между тарелками, мм Давление, МПа Температура, °С
1800
500
0,59
верх
сеченне подачи питания
низ
Кратность орошения
48
61
106
3:1
Примечание. В каждой колонне 60 Клапанных двухпоточных тарелок.
того что температура низа колонн И и 14 относительно невелика {табл. 41), их обогрев осуществляется паровыми рибойлерами 13.
Описываемая установка имеет проектную мощность 417 тыс. т в год, в том числе 257 тыс. т нестабильного бензина и 160 тыс. т жирного газа. В процессе эксплуатации производительность установки превысила проектную. Чистота пропан-пропиленовой фракции 96%, бутан-бутиленовой 97%; отбор от потенциала соответственно 82 и 95%; сухой газ содержал всего 0,3% фракции С4 и почти на 90% состоял из фракций до С 2 (включительно).
Обычно на блоке ректификации непредельных газов практикуется выделение фракций С3 и С4 без их последующего разделения на предельную и непредельную часть. Если на НПЗ предусмотрены полимеризация пропилена или использование его в качестве компонента сырья алкилирования, сопутствующий пропилену пропан не оказывает вредного влияния на эти процессы. Поскольку пропилен нацело вступает в реакцию, пропан легко выделить затем из продуктов. То же можно сказать и о н-бутане. Если на заводе существует установка каталитического крекинга, ей обычно сопутствует установка алкилирования изобутана олефина-ми; балластной фракцией в этом процессе является н-бутан, который выделяют затем из катализата.
ИСПОЛЬЗОВАНИЕ КОМПОНЕНТОВ ГАЗА
С установок АГФУ блока разделения непредельных газов уходят сухой газ, пропан-пропиленовая и бутан-бутиленовая фракции. В типичных заводских газах из непредельных углеводородов присутствуют только олефины: этилен, пропилен, бутилены. Углеводороды более высокой непредельности — ацетилен, бутадиен — содержатся лишь в газах пиролиза, а в газах термического крекинга появляются только при значительном ужесточении режима.
Полимеризацией газообразных олефинов можно получить разнообразнейшие продукты — от легких бензиновых фракций до высокомолекулярных полимеров, молекулярная масса которых достигает двух-трех миллионов.
В 30-х годах процесс селективной каталитической полимеризации бутиленов широко использовали с целью последующего гидрирования димера (ызо-CgHie) и получения таким образом технического изооктана — компонента авиационного бензина. Процесс этот впоследствии потерял свое значение, так как был вытеснен каталитическим алкилированием бутиленами изобутана, содержащегося в больших количествах в газах каталитического крекинга. Позднее был внедрен процесс получения полимер-бензина на основе пропилена, который был менее дефицитен. В качестве катализатора используют фосфорную кислоту, нанесенную на кварц. Полимеризацию проводят при 220—230 °С, 6,5—7,0 МПа и объемной скорости подачи сырья от 1,7 до 2,9 ч-1. Применяется и совместная полимеризация пропиленов и бутиленов или бутиленов и амиленов.
Насыщенные углеводороды, содержащиеся в сырье полимеризации, естественно, не вступают в реакцию, но оказывают благоприятное влияние на тепловой баланс реактора, препятствуя чрезмерно глубокому протеканию реакции, сопровождающейся образованием более тяжелых полимеров. Теплота полимеризации составляет «1550 кДж (370 ккал) на 1 кг пропилена. Максимальным октановым числом — около 90 — обладает полимеризат, полученный из бутиленовой фракции (димер); бензин — продукт полимеризации пропилена •— имеет октановое число примерно на
10 ниже (80—82 по моторному методу). По химическому составу полимер-бензин, естественно, состоит почти нацело из олефинов, что обусловливает его невысокую химическую стабильность при хранении и низкую приемистость к этиловой жидкости; при добавке 3 мл ТЭС октановое число полимер-бензина повышается всего на 3—4 единицы.
Большая потребность нефтехимической промышленности в пропилене заставила отказаться от использования этого олефина для производства полимер-бензина.
КАТАЛИТИЧЕСКОЕ АЛКИЛИРОВАНИЕ ИЗОБУТАНА ОЛЕФИНАМИ
Сущность процесса. Процесс алкилирования заключается в присо-" единении олефина к парафину с образованием соответствующего углеводорода более высокой молекулярной массы. С точки зрения строения молекулы, образовавшийся алкилпарафин можно рассматривать как исходный парафин, у которого один атом водорода заменен алкильной группой. Однако основная реакция сопровождается рядом побочных, в результате чего образуется более или менее сложная углеводородная смесь.
В нефтеперерабатывающей промышленности были осуществлены различные модификации процесса алкилирования. Наиболее распространены установки для алкилирования изобутана олефи-нами (в основном бутиленами) с получением широкой бензиновой фракции — алкилата. Алкилат, состоящий почти нацело из изопарафинов, имеет высокое октановое число (90—95 по моторному методу) и применяется в качестве компонента автомобильных и авиационных бензинов. Некоторое время в качестве высокооктанового компонента авиационных бензинов широко использовали также продукт алкилирования бензола пропиленом — изопропил-бензол (кумол). В связи с непрерывным сокращением производства авиационного топлива для карбюраторных двигателей кумол утратил свое значение как топливный компонент, но используется как полупродукт при производстве фенола и ацетона. В годы
II мировой войны вырабатывали (в ограниченном количестве) еще один высокооктановый компонент—неогексан (2,2-диметил-бутан)—путем термического алкилирования изобутана этиленом.
В 1932 г. В. Н. Ипатьев показал возможность взаимодействия изобутана, считавшегося до того «инертным» углеводородом, с оле-финами. В качестве катализатора был использован А1СЬ. Эта реакция, разработанная затем с применением других катализаторов — серной кислоты и позднее фтористого водорода, — была быстро внедрена в промышленность. Первые промышленные установки сернокислотного алкилирования были введены в эксплуатацию в конце 30-х годов, а установки фтористоводородного алкилирования в 1942 г. Целевым продуктом вначале был исключительно компонент авиационного высокооктанового бензина, и лишь в послевоенные годы алкилирование стали использовать для улучшения моторных качеств товарных автомобильных бензинов.
При промышленном процессе алкилирования получать высокооктановый компонент бензина проще и дешевле, чем в применяемом ранее процессе каталитической полимеризации бутиленов с последующим гидрированием димера в изооктан. Замена селективной полимеризации бутиленов каталитическим алкилирова-нием изобутана бутиленами давала следующие преимущества:
1) получение бензина, богатого изооктаном, в одну ступень вместо двухступенчатого процесса полимеризация — гидрирование;
2) вдвое меньший расход ценных олефинов на получение одного и того же количества высокооктанового компонента;
3) отсутствие расхода водорода для гидрирования;
4) более полное вовлечение олефинов, содержащихся в заводских газах; при алкилировании олефины вступают в реакцию нацело, тогда как при полимеризации менее активный олефин (например, н-бутилен при полимеризации смеси бутиленов) остается частично непрореагировавшим.
Однако каталитическое алкилирование изобутана начало усиленно развиваться лишь вследствие широкого внедрения установок каталитического крекинга. Газ каталитического крекинга, богатый изобутаном, обеспечил установки алкилирования одним из компонентов сырья, а для получения олефинов приходилось использовать и газы термических процессов.
Основные факторы процесса. В качестве промышленных катализаторов алкилирования применяют только серную кислоту и жидкий фтористый водород. Выбор этих веществ обусловлен их хорошей избирательностью, удобством обращения с жидким катализатором, относительной дешевизной, продолжительными циклами работы установок благодаря возможности регенерации или непрерывного восполнения активности катализатора.
Каталитическому алкилированию в присутствии серной кислоты или фтористого водорода можно подвергать только парафины изостроения, содержащие активный третичный атом углеводорода. При этом алкилирование изобутана этиленом идет с трудом, очевидно, вследствие стабильности образующихся промежуточных соединений — эфиров. Алкилирование пропиленом и особенно бу-тиленами протекает достаточно глубоко. Решающее значение имеет концентрация кислоты. Так, для алкилирования изобутана бутиленами можно использовать 96—98%-ную серную кислоту, для алкилирования же пропиленом применяют только 98— 100%-ную кислоту.
Характерно, что в результате основной реакции присоединения изобутана к олефину-происходит одновременная структурная изомеризация, что свидетельствует о наибольшей вероятности кар-боний-ионного цепного механизма. Наряду с основной реакцией алкилирования, при которой на 1 моль изобутана расходуется 1 моль олефина, протекают побочные реакции.
1. Перенос водорода, или самоалкилирование. Так, взаимодействие изобутана с пропиленом частично идет в следующем направлении:
2изо-С4Н10 -j- C3He -> изо-С8Н18 -j- С3Н8
Эта реакция нежелательна, так как вызывает повышенный расход изопарафина и образование малоценного пропана.
2. Деструктивное алкилирование. Первичные продукты алкилирования расщепляются, и образующийся олефин (отличающийся от исходного) вновь реагирует с исходным парафином, например:
2«зо-С4Н10 + СдН, -> изо-С5Н12 + изо-СвН14
3. Полимеризация. Кислотные катализаторы вызывают полимеризацию олефинов, поэтому неблагоприятный для алкилирования режим — малая концентрация изопарафина, недостаточная активность катализатора и повышенная температура — вызывают появление полимеров в составе продуктов алкилирования.
В процессе алкилирования происходит постепенное дезактирование катализатора — падение концентрации кислоты и ее потемнение, вызываемые взаимодействием кислоты с непредельными углеводородами и влагой. Влага может содержаться в сырье, а также образуется в результате побочного взаимодействия олефинов с. кислотой:
СлН2П +
HaS04< -¦?-
СЛН2Л_2 + 2Н20 -t-
so2
При понижении концентрации кислоты ослабляется целевая реакция алкилирования и увеличивается доля полимеризуклцихся олефинов. Требуемую концентрацию кислоты в реакционной зон поддерживают путем частичной или полной замены отработанной кислоты свежей.
Реакция алкилирования протекает с положительным тепловым эффектом («960 кДж, или 230 ккал на 1 кг алкилата). Дл поддержания изотермического режима выделяющееся тепло необ ходимо непрерывно отводить из реакционной зоны.
Термодинамически алкилирование — низкотемпературная реак ция. Пределы температур промышленного сернокислотного алки дирования от 0 до 10 °С; алкилирование в присутствии фтористого водорода проводят при несколько более высокой температуре — примерно 25—30 °С. Такое различие объясняется тем, что при температурах выше 10—15°С серная кислота начинает интенсивно окислять углеводороды.
Понижение температуры хотя и замедляет алкилирование, но увеличивает его избирательность в сторону образования первичного продукта алкилирования, в связи с чем качество получаемого алкилата улучшается. Снижение температуры на 10—11 °С вызывает повышение октанового числа алкилата примерно на 1. Чрезмерное понижение температуры ограничено температурой затвердевания кислоты-катализатора, а также увеличением вязкости катализатора и, следовательно, трудностью его диспергирования в реакционной смеси. Возможность проведения реакции при более высокой температуре — одно из достоинств фтористого водорода, так как это упрощает систему отвода тепла от реакционной смеси.
Давление в реакторе выбирают с таким расчетом, чтобы все • углеводородное сырье или основная его часть находилась в жидкой фазе. Давление в промышленных реакторах составляет в среднем 0,3—1,2 МПа.
Применяемые катализаторы вызывают полимеризацию олефинов, поэтому необходимо, чтобы концентрация олефинов в реакционной смеси была значительно ниже, чем требуется по стехиометрическому уравнению реакции. С этой целью практикуется раз-
Таблица 42. Показатели и выход продукта при производстве алкилата — компонента автомобильного бензина
Данные О. Иверсона и Л. Шмерлинга
| Олефиновое сырье | |||
| Показатели |
пропилен | бутнлены | амилены |
|
Расход изобутана, кг на 1 кг олефинового сырья стехиометрический |
1,38 | 1,035 | 0,83 |
| фактический |
1,38—1,47 | 1,035—1,096 |
0,83—1,20 |
| Выход суммарного ал | 1,75—1,78 |
1,70—1,72 | 1,6* |
|
килата, объем на - 1 объем олефинового сырья Октановое число алкилата без ТЭС | 89—91 (и. м.) | 92—96 (и. м.) | 88—90 (и. м.) |
| с 0,8 мл ТЭС иа 1 л | 87—90 (м. м.) 100—104 (и. м.) | 92—94 (м. м.) 103—109 (и. м.) | 87—89 (м. м.) 100-102 (и. м.) |
| г | 39—101 (и. м.) | 100—104 (м. м.) | 98—100 (м. м.) |
* Выход депентанизированного суммарного алкилата.
бавление сырья потоком изобутана, непрерывно циркулирующего в системе. Мольное соотношение изобутан : олефин в углеводородной смеси, поступающей на алкилирование, составляет обычно (4-=-10) : 1; наиболее часто применяется шести- или семикратное разбавление. При избытке изобутана повышается качество алкилата и подавляются не только полимеризация, но и побочные реакции деалкилирования. Так как при большой кратности изобутана избирательность процесса увеличивается, расход олефинов на единицу количества изобутана сокращается. Увеличивать соотношение изобутан : олефин более 10 : 1 малоэффективно. Следует учитывать, что при повышенной кратности изобутана возрастают эксплуатационные расходы на его циркуляцию и охлаждение, а также требуется увеличивать размеры основных аппаратов.
Большое значение имеет интенсивность перемешивания углеводородной фазы и катализатора, в связи с тем что взаимная растворимость их очень невелика. Очевидно, реакция идет в катали-заторной фазе и на границе раздела фаз между растворенными в катализаторе изобутаном и олефиновым компонентом сырья. , В отсутствие или при недостатке изобутана контакт олефина с кислотой вызывает полимеризацию олефинов. Интенсивное перемешивание способствует также отделению образовавшегося алкилата от катализатора. Стремление увеличить концентрацию изобутана в месте ввода смеси привело к разработке специальных смесительных и циркуляционных устройств, позволяющих увеличивать со-
отношение изобутана и олефина в поступающей смеси до 100: lH и более. Из данных табл. 42 видно, что соотношение между И30-Н бутаном и олефином в исходной сырьевой смеси должно бытьЯ близко к теоретическому. Наиболее высокое октановое число алки-Я лата наблюдается при бутиленовом сырье. Я
Понятие продолжительности реакции является для ДанногоЯ процесса условным, так как, в соответствии с изложенным выше,И реакция может протекать не во всем объеме катализатора.. Отно-В сительными являются и принимаемый обычно за основу Шоказа-В тель объемной скорости и обратная ему величина — УСловнаяВ продолжительность реакции. В
За объем катализатора должен быть принят объем кислоты,В диспергированной в реакторе, так как остальная ее часть,, Попа-В дающая в зону отстоя или не образовавшая эмульсии и3(-за не-В достаточно интенсивного перемешивания, фактически не; будетВ катализировать алкилирование. Однако учесть этот объем невоз-В можно, и в данном случае условная объемная скорость выражает-В ся объемным количеством олефинов, подаваемым в час на едининВ цу объема катализатора. Объемная скорость в значительнойВ степени зависит от интенсивности перемешивания реакцноннояВ массы, особенно в местах ввода олефинов. Недостаточный массо^| обмен вызывает местные перегревы реакционной смеси и сниже-В ние качества алкилата. Средняя объемная скорость подачи олефи-^| нов для сернокислотного алкилирования 0,1—0,6 ч-1.
Полнота протекания реакции обеспечивается при длительносп^В пребывания углеводородной фазы в реакторе 5—10 мин для фто^И ристоводородного алкилирования и 20—30 мин для сернокислотно^В го алкилирования. При этом объемное соотношение каталшзаторЯИ углеводород принимают равным 1 : 1 (это установлено исходя из i наличия в реакторе однородной эмульсии углеводородов в кисло- i те). Увеличение относительного объема кислоты не вредит процес- ! су, но увеличивает вязкость смеси и соответственно расход энер- ! гии на перемешивание; уменьшение доли кислоты приводит к об- ! разованию ее эмульсии в углеводороде, к ухудшению Качества I алкилата и увеличению расхода катализатора. Соотношение кис- I лота: углеводород несколько изменяется в зависимости от кон- ? центрации кислоты, ее плотности, качества сырья, типа реактора \ и др. Указанное выше соотношение 1 : 1 является усредненным. I Промышленные установки сернокислотного алкилирования.^ В нефтеперерабатывающей промышленности наиболее распростра(Я нен процесс сернокислотного алкилирования. В зависимости о^| конструкции реактора и системы погоноразделения возможны не|В сколько вариантов технологической схемы установки. Выще отме^Ч чалось, что реакция алкилирования протекает со значительным положительным тепловым эффектом. Выделяющееся тепло отводят двумя способами: 1) охлаждением реакционной смеси через теплообменную поверхность; 2) охлаждением смеси частичным ее испарением. Соответственно имеется два типа реакторов.
На рис. 105 и 106 представлены эскизы реакторов первого типа, так называемых контакторов.
На рис. 105 изображен вертикальный контактор (общая высота «11,7 м, внутренний диаметр «2 м) более старого типа, рассчитанный на небольшую пропускную способность. Реакционную смесь охлаждают аммиаком или пропаном, циркулирующим через двойные трубки.
Хмдоагент 1
Хладоагент
Продукты
реакции
Кислота
Сырье
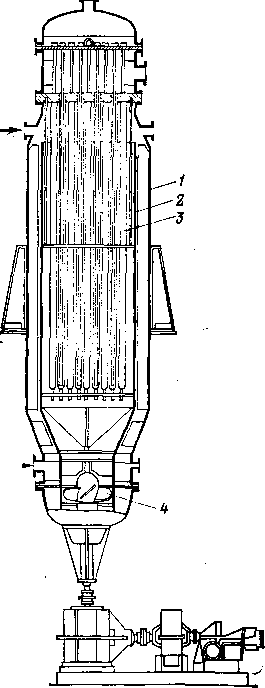
Рис. 105. Вертикальный контактор:
I — корпус; 2 — цилиндрический кожух; 3 — трубный пучок; 4--пропеллерный насос.
Выйдя через открытые концы внутренних трубок, сжиженный газ переходит в наружный кольцевой зазор и, испаряясь, выходит из системы. Отвод тепла регулируют, изменяя давление в системе охлаждения. Реакционную смесь перемешивают пропеллерным насосом; в качестве привода служит электродвигатель или паровая турбина. Рабочий объем реактора разделен цилиндрической перегородкой; смесь углеводородов и кислоты, приводимая в движение пропеллерным насосом, непрерывно циркулирует в аппарате, поднимаясь по кольцевому сечению и опускаясь по внутреннему цилиндру, где от нее отнимается тепло через поверхность охлаждающих трубок. Для упорядочения восходящего потока к цилиндрической перегородке приварены вертикальные ребра.
В горизонтальном контакторе (рис. 106) более удачно осуществлен ввод сырья и катализатора— они попадают сразу в зону наиболее интенсивного смешения. Далее смесь прокачивается по кольцевому пространству и в противоположном конце аппарата поворачивает во внутренний цилиндр. Горизонтальное расположение аппарата устра-
няет необходимость в зубчатой передаче к приводу и облегчает обслуживание контактора. В аппарате происходит чрезвычайно интенсивная циркуляция; кратность ее достигает на крупных установках 200 м3 в мин. При такой кратности циркуляции поступающая смесь практически мгновенно смешивается с эмульсией, заполняющей реактор. Соотношение изобутан: олефин в месте поступления сырьевого потока достигает 500:1 и более. Горизон-
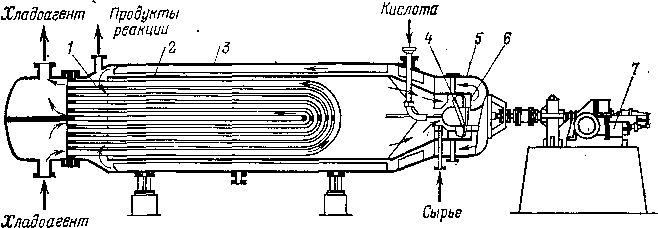
Рис. 106. Горизонтальный контактор:
1 — трубный пучок; 2, 5 — циркуляционная труба; 3 — корпус; 4 — пропеллерная мешалка:
6 — направляющие лопасти; 7 — турбина.
тальные контакторы конструктивно проще. Они отличаются также тем, что в качестве хладоагента используется поток продуктов реакции. Емкость их больше, чем у вертикальных аппаратов, но ее можно увеличивать лишь до определенных пределов, так как применение очень крупных контакторов ухудшает качество смешения; поэтому предпочитают устанавливать не менее трех-четы-рех контакторов.
Большое значение имеет система питания аппаратов. Опыт эксплуатации установок сернокислотного алкилирования показал, что циркулирующие изобутан и катализатор целесообразно подавать в контактор последовательно, а исходную углеводородную смесь (изобутан и олефины) лучше подавать параллельно, распределяя ее на потоки по числу контакторов. При этом относительная доля олефинов в реакционной смеси уменьшается, что позволяет повысить селективность процесса, сократить расход серной кислоты и улучшить качество алкилата.
Подобное изменение системы питания реакторов на одной из установок алкилирования грозненского завода снизило* расход кислоты на 35%. Иногда при такой схеме эксплуатации контакторов практикуется дополнительная параллельная подача кислоты. Например, систему из четырех контакторов циркулирующий изобутан проходит последовательно, исходный углеводородный
поток делится на четыре параллельных, а кислота, пройдя первый и второй реакторы, отстаивается от углеводородной фазы в отстойнике и вновь возвращается в первый реактор. Аналогично этому, в третий реактор поступает кислота из отстойника, обслуживающего третий и четвертый реакторы.
В реакторах алкилирова- пония большое значение имеет интенсивность перемешивания реакционной смеси. Снижение Е 91 температуры повышает окта- ~
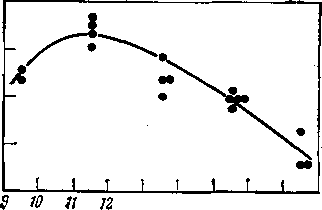
13 п я Ш П id Температура°?
Рис. 107. Влияние температуры на октановое число алкилата.
новое число алкилата <§ 30
Ч.
&
(рис. 107). Однако было показано, что для данного числа J 89 оборотов (320—380 в минуту) температура не должна быть ниже 10—11°С, так как при дальнейшем ее снижении вязкость реакционной эмульсии повышается настолько, что требуется большее число оборотов мешалки. Таким образом, температура реакции и число оборотов мешалки должны находиться в оптимальном сочетании,-в частности для вертикальных контакторов рекомендованы 8 °С -и 500—520 оборотов в минуту при времени контакта 8—10 мин*.
Современным заводам высокой мощности наиболее полно отвечает каскадный реактор (рис. 108). Это реактор второго типа, где охлаждение смеси идет за счет ее частичного испарения. Каскадный реактор — горизонтальный аппарат цилиндрической формы с несколькими секциями смещения, снабженными мешалками, и двухсекционной зоной отстаивания. Циркулирующий изобутан и серная кислота поступают в первую секцию смешения; исходное сырье — смесь изобутана с олефинами — равномерно распределяется по всем секциям, благодаря чему в каждой зоне обеспечен значительный избыток изобутана. Схема смесительной секции показана на рис. 109. Над мешалками размещены змеевики для ввода сырья и вертикальные перфорированные трубы для циркуляции эмульсии. Направление потока эмульсии видно на рис. 109.
Принятый режим давления в реакторе такой: в первой секции смешения 0,15—0,20 МПа, перепад давления на каждую секцию 0,01—0,02 МПа; средняя объемная скорость подачи олефинов составляет примерно 0,3 ч-1. В двух последних секциях кислота отделяется от углеводородного слоя. Температура и давление в реакторе обеспечивают частичное испарение углеводородной фазы, в основном наиболее легкого компонента — изобутана. Испарив-
шийся газ отсасывают компрессором и после охлаждения и конденсации возвращают в реакционную зону. Выделяющееся тепло реакции снимается за счет тепла испарения изобутана. Температура в реакторе поддерживается на заданном уровне автоматически.
В каскадном реакторе может быть от трех до шести секций смешения. Существуют установки с реактором, в котором имеется шесть секций смешения (по три с каждой стороны) и одна зона отстаивания, расположенная в средней части аппарата. На одной из крупнейших установок сернокислотного алкилирования производительностью до 950 м3 алкилата в сутки установлено два
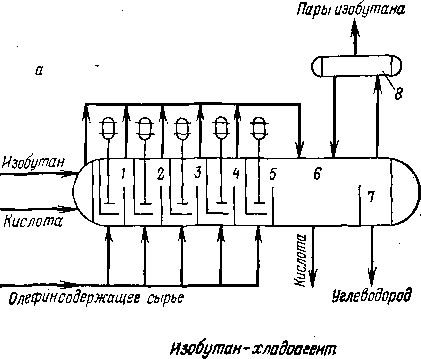
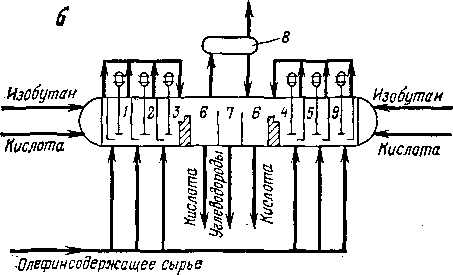
Рис. 108. Горизонтальные реакторы каскадного типа: а — пятисекцнонный; б — сдвоенный;
1, 2, 3, 4, 5 — секции; б — зона отстаивания кислоты; 7 — зона вывода алкилата; 8 — емкость нзобутана.
5-ступенчатых реактора диаметром «3,5 м и длиной «22 м.
Наличие каскадных реакторов, работающих по принципу «автоохлаждения», упрощает и удешевляет установки алкилирования, так как позволяет отказаться от хладоагента (аммиак, пропан). Сопоставление удельных расходов серной кислоты в реакторах описанных конструкций свидетельствует о преимуществах каскадного реактора; для вертикального контактора этот расход дости-
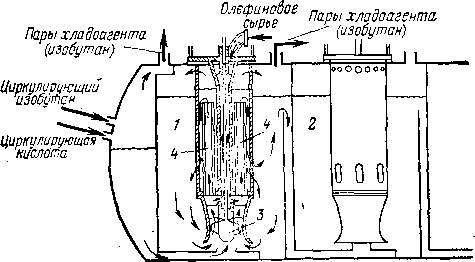
Рис. 109. Смесительная секция каскадного реактора:
1, 2 — секции реактора; 3 — мешалка; 4 — циркуляционные трубы.
гает 200—250 кг на 1 т алкилата, в каскадном 60—100 кг/т. Октановое число целевого продукта (легкий алкилат) в первом случае 90—91 (по моторному методу), во втором 92—95. Однако каскадные реакторы имеют некоторые недостатки: секции взаимосвязаны, и нарушение режима в одной из них может привести к разлажива-нию работы аппарата в целом; по ходу движения эмульсии концентрация изобутана снижается.
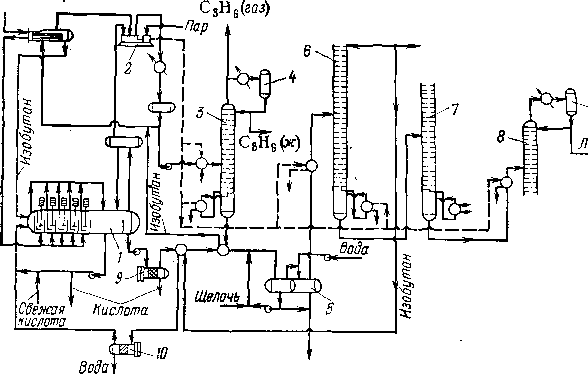
Принципиальная технологическая схема установки сернокислотного алкилирования представлена на рис. 110. Эта схема характеризуется сложным блоком погоноразделения, состоящим из четырех ректификационных колонн: пропановой, изобутановой,
бутановой и колонны вторичной перегонки алкилата. Исходная углеводородная смесь охлаждается испаряющимся бутаном в холодильнике и поступает пятью параллельными потоками в смесительные секции реактора в первую секцию подают также циркулирующий изобутан и серную кислоту. Из отстойной зоны реактора выходят серная кислота (на циркуляцию или на сброс) к углеводородная смесь, которая проходит нейтрализацию щелочью и промывку водой.
Испарившаяся в реакторе часть углеводородов через каплеог-бойник поступает на прием компрессора 2, который подает ее через холодильник в емкость и пропановую колонну 3. Эта колонна служит для отделения и вывода из системы пропана во избежание его постепенного накапливания в системе. Остаток пропано-вой колонны — изобутан — частично циркулирует через сырьевой холодильник и прием компрессора 2, а частично присоединяется к общему потоку циркулирующего изобутана. Основной углеводородный поток из отстойника 5 направляется в изобутановую колонну 6 для отделения рециркулирующего изобутана. Головной логон этой колонны — изобутан — возвращают в первую смеси-
ИсхоЗные
углевсЗороды
"4пЮ
Т.д-^ Тяжелый
алкилат
Щелочь+бода
Рис. 110. Технологическая схема сернокислотного алкилирования нзобутана олефинами:
1 — реактор; 2 — компрессор; 3— пропановая колонна; 4 —емкости орошения; 5 — отстойник; 6 — изобутановая колонна; 7 — бутановая колонна; 8 — колонна вторичной перегонки алкнла» та; 9— коалесцнрующнй аппарат; 10— сепаратор.
тельную секцию реактора. При некотором избытке свежего изобутана в исходном сырье предусмотрено его удаление. Остаток изо-бутановой колонны поступает на дальнейшее разделение в бутано-вую колонну 7, а остаток бутановой колонны — в колонну 8 для перегонки алкилата. С верха этой колонны уходят пары целевой фракции (легкий алкилат), а с низа — тяжелый алкилат, выкипающий выше 150—170 °С и используемый обычно как компонент .керосина.
В табл. 43 представлены данные о ректификационных колоннах крупной установки алкилирования и о режиме их работы. Для четкого разделения продуктов колонны снабжены паровыми ри-бойлерами и, как видно из таблицы, имеют значительное числа тарелок. Вместо потока циркулирующего изобутана можно подвергать депропанизации отгон изобутановой колонны. Достоинством такой системы является несколько более высокая концентрация пропана в сырье пропановой колонны, что облегчает выделе-ние пропана.
На многих современных установках сернокислотного алкилирования углеводородный поток, выходящий из реактора, очищают бокситом и лишь затем нейтрализуют щелочью и промывают водой. Такая очистка нужна для отделения сложных эфиров, образующихся под действием катализатора. При обработке щелочью нейтрализуется только часть кислотных продуктов, а наиболее стойкие из сложных эфиров либо разлагаются при нагреве и вызывают постепенное шламообразование в системе погоноразделе-ния и коррозию, либо попадают в товарный алкилат и снижают его антидетонационные показатели.
Для удаления указанных вредных примесей углеводородный поток после реактора направляют последовательно в коалесци-рующий аппарат, заполненный стеклянной ватой, и в одну-две колонны, заполненные бокситом. Назначение коалесцирующег» аппарата — удаление мельчайших капелек кислоты, содержащихся в углеводородном потоке. Бокситовые колонны работают попеременно: через 1 кг боксита можно пропустить от 500 до 1500 ма алкилата (в зависимости от степени его загрязненности эфирами), после этого углеводородный поток переключают на вторую колонну. Удаление бокситом следов серной кислоты и эфиров основано на избирательной адсорбции этих полярных соединений. О сработанности боксита судят по началу десорбции — выделения вместе с алкилатом нейтральных сложных эфиров, содержащих серу. Загрязненный боксит продувают водяным паром, промывают
Таблица 43. Размеры и технологические параметры ректификационных колонн на установке сернокислотного алкилирования
Данные В. П. Суханова
|
Показатели | Изо- бутановая колонна |
Бутановая колонна |
Пропановая колонна |
Колонна вторичной перегонки алкилата |
| Диаметр, мм | 3000 | 1600 |
1600 • | 1800 |
|
Высота, мм | 50085 |
26500 | 27400 | 19062 |
| Число тарелок |
80 | 40 | 40 |
20 |
| Избыточное давление, МПа Температура, °С |
0,5—0,6 | 0,35—0,40 |
1,6—1,7 | к 0,03 |
|
, верх | 45—55 |
45—50 | 20—45 | 100—115 |
| низ Состав ректификата, % (об.) | 95—120 |
125-135 | 95—100 |
До 220- |
| Сз |
2,0 | — | 95,0 |
— |
| ЫЗ0-С4НЮ |
89,0 | 2,8 | 5,0 | — |
| Н-С4Н10 | 9,0 | 71,0 |
— | — |
| С5 и выше | - | 26,2 |
— | 100 |
водой и осушают, пропуская через него углеводородный газ. Очи* щенный бокситом углеводородный поток подвергают щелочной и водной промывке. Благодаря тому, что в схему алкилирования вводят бокситную очистку, продолжительность непрерывного пробега установки увеличивается и превышает 8 месяцев.
Решающее влияние на образование эфиров оказывает интенсивность перемешивания эмульсии в реакторе; при высокой интенсивности перемешивания эфиры разлагаются. Анализ работы промышленных установок показал, что минимальное содержание эфиров в суммарном алкилате наблюдалось при концентрации серной кислоты 89— 92% (масс.) (рис. 111). Наличие минимума объясняется тем, что при более высокой концентрации кислоты усиливается ее взаимодействие с олефинами, т. е. активизируется и образование эфиров. При чрезмерно низких концентрациях кислоты селективность ее как катализатора алкилирования падает, и эфиры уходят с продуктами алкилирования.
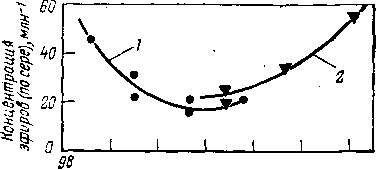
96 94 92 90 88 8$
Концентрация H2S04,% (масс.)
Рис. 111. Зависимость содержания эфиров в суммарном алкилате от концентрации серной кислоты:
1 — алкилирование изобутана бутиленом; 2 — ал-килировзние изобутана пропиленом.
Большое значение имеют чистота и отсутствие влаги в сырье алкилирования. Было показано, что при снижении влагосодержа-ния сырья от 0,03 до 0,001% (масс.) можно сократить расход серной кислоты на 16 кг (на 1 т алкилата). Сочетание отстойников, поставленных на потоке охлажденного сырья перед реакторами, с' адсорбционным удалением влаги должно дать значительный экономический эффект.
В результате побочных реакций алкилат, как правило, содержит более или менее тяжелые фракции — выкипающие выше 170 °С, т. е. выше температуры конца кипения товарных бензинов. В связи с этим необходима колонна вторичной перегонки алкилата. Выход тяжелого алкилата при нормальном режиме не превышает 5%. Решающее значение имеет при этом концентрация изобутана в реакционной зоне. Поскольку результат реакции определяется составом потока, выходящего из реактора, важно, чтобы высокая концентрация изобутана сохранялась и в этом потоке.
Присутствие инертных разбавителей (н-бутан и пропан) ухудшают качество алкилата, и чистота применяемого для циркуляции изобутана играет весьма большую роль. Так, при прочих равных режимных показателях увеличение концентрации изобутана в от-
ходящем из реактора углеводородном потоке с 40 до 70% (об.) вызвало повышение октанового числа суммарного алкилата с 90 до 92,8; при этом в первом случае разность октановых чисел суммарного и легкого алкилата составила «0,9, а во втором она была всего 0,3, что свидетельствует о приближении выхода легкого алкилата к 100%.
Недостатком сернокислотного алкилирования является довольно значительный расход серной кислоты вследствие разбавления ее побочными продуктами реакции. Наименьший расход кислоты наблюдается, если в качестве олефинового сырья применяют чистые бутилены; при использовании пропилена расход кислоты увеличивается примерно втрое. Как было показано выше, расход кислоты связан также с интенсивностью перемешивания реакционной смеси и с температурой, повышение которой увеличивает степень разбавления кислоты. Увеличивается расход кислоты и при наличии в сырье таких примесей, как сернистые соединения и влага. Затраты на катализатор можно снизить при использовании отработанной кислоты для иных целей (например, для очистки масел и других нефтепродуктов), а также при ее регенерации.
Промышленные установки фтористоводородного алкилирова-яия. На зарубежных заводах довольно широко распространены установки алкилирования с фтористым водородом как катализатором. Жидкий фтористый водород по сравнению с серной кислотой более активен и благодаря его летучести (т. кип. 20 °С) легче регенерируется. Еще одним достоинством этого катализатора является более низкая плотность (?»1,0 против 1,84 для серной кислоты). Это облегчает образование эмульсии катализатора с углеводородной фазой в реакторе и даже позволяет отказаться от механического перемешивания. Концентрация применяемого катализатора «90%, и она относительно мало влияет на выход и качество алкилата. Однако система регенерации катализатора довольно сложна.
На рис. 112 представлена принципиальная технологическая схема установки фтористоводородного алкилирования. Исходное сырье проходит бокситную осушку в колоннах 1 и поступает в реакторы 2. Применяют реакторы трубчатого типа с водяным охлаж-' дением, так как реакция протекает при 20—40 °С. На некоторых установках реакторы конструктивно объединены с отстойниками. Особенность установок фтористоводородного алкилирования — наличие системы регенерации катализатора. Алкилат после отстаивания от основного объема HF поступает в колонну-регенератор 4, где циркулирующий изобутан отделяется в виде бокового погона. Колонна-регенератор 4 обогревается внизу посредством циркуляции остатка через печь 3. При этом от алкилата отпариваются изобутан, пропан и катализатор. При. нагреве остатка до 200— 205 °С разрушаются также органические фториды, образующиеся
в качестве побочных продуктов реакции. С верха колонны-регенератора 4 уходят в газовой фазе пропан, фтористый водород и некоторое количество изобутана. После конденсации часть этой смеси возвращают в реакторы, часть подают на орошение колонны 4, а остальное направляют в пропановую колонну 6, с верха
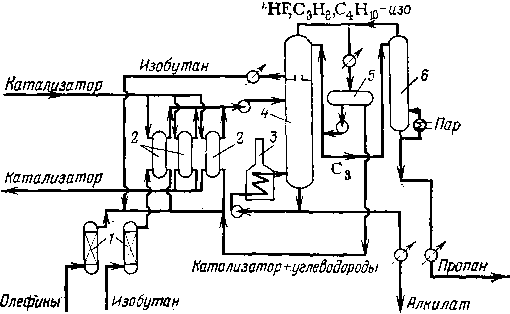
Рис. 112. Схема установки алкилирования изобутана олефинами в присутствии фтористого водорода:
1 — осушительные колонны; 2 — реакторы; 3 — печь; 4 — колонна-регенератор; 5 — отстойник; 6 — пропановая колонна; 7 — паровой нагреватель. .
которой уходит отпаренный фтористый водород, а с низа—пропан
со следами изобутана.
Для более полного возврата катализатора предусмотрена также регенерация (в отдельном блоке) части кислотного слоя из отстойника. Алкилат с низа колонны 4 после охлаждения проходит через бокситные колонны, где освобождается от остатка фтористых соединений. В результате хорошей регенерации расход катализатора не превышает 1 кг на 1 т алкилата.
Применение фтористоводородного катализатора вследствие его токсичности и значительной летучести требует соблюдения строгих мер предосторожности. Осуществляется непрерывный автоматический контроль за точками возможной утечки фтористого водорода: в потоках воды, охлаждающей реакторы и конденсаторы, в холодильниках кислоты и др. Зона, где размещены кислотные насосы и аппараты, содержащие кислоту, считается опасной, и в нее можно входить только в специальных кислотоупорных костюмах и масках. Большое внимание уделяется подбору материалов и конструкций аппаратуры, оборудования и трубопроводов. Применяют специальные прокладочные материалы из стойких к HF веществ — фторорганических пластмасс. В местах наибольшей коррозии используют монель-металл325, а основную аппаратуру изготовляют из углеродистой стали.
На нефтеперерабатывающих заводах США преобладает сернокислотное алкилирование, однако к началу 1977 г. доля фтористоводородного алкилирования достигала 326 уже 40% от общего (по сырью) против 30,6% в 1970 г. Относительно более быстрый рост мощностей установок алкилирования с фтористым водородом объясняется совершенствованием схемы процесса. Например, на некоторых установках, где в качестве олефинов используют только бутилены, из схемы исключены колонны отпарки HF и пропана, а отгон изобутановой колонны непосредственно возвращают в процесс. Описаны установки, где смешение сырья с кислотой осуществляют в стояке (вертикальные трубы большого диаметра, соединяющие выходные штуцеры кислотных охладителей с входным штуцером реактора). При этом сами реакторы лишены перемешивающих устройств, что устраняет разрушение аппарата от корродирующего действия фтористого водорода.
Увеличение ресурсов сырья для алкилирования. Ресурсы сырья для алкилирования ограничены. Изобутан содержится в значительной концентрации лишь в газах каталитического крекинга и гидрокрекинга; он может быть выделен также из попутного газа. Бутилены содержатся в газах каталитического, термического крекинга и коксования и отсутствуют в газах, получаемых при гидро-генизационных процессах.
Ресурсы изобутана можно увеличить путем изомеризации н-бутана на катализаторах, родственных катализаторам изомеризации углеводородов С5—Сб. Установка изомеризации н-бутана может быть скомбинирована с установкой алкилирования — с общ%р изобутановой колонной.
Для расширения ресурсов олефинов в процесс алкилирования вовлекают пропиленовую фракцию или подвергают дегидрированию н-бутан. Однако, с одной стороны, алкилат на основе пропилена или смеси его с бутиленами имеет более низкое октановое число: при использовании только пропилена — примерно на 5 единиц. С другой стороны, пропилен является ценным нефтехимическим сырьем, а дегидрирование м-бутана чаще проводят с целью получения бутадиена — сырья для производства синтетического каучука. Возможно, что ресурсы олефинов С3—С4 увеличатся за счет возрастающей тенденции к утяжелению сырья пиролиза И ужесточению режима установок каталитического крекинга.
ИСПОЛЬЗОВАНИЕ СЕРОВОДОРОДА, СОДЕРЖАЩЕГОСЯ В ЗАВОДСКИХ УГЛЕВОДОРОДНЫХ ГАЗАХ
Углеводородные газы заводов, перерабатывающих сернистые неф ти, содержат сероводород. Часть этого сероводорода образуете при термической или термокаталитической деструкции наимене стабильных сернистых соединений, содержащихся в нефтяном сырье, при его термическом и каталитическом крекинге и коксовании. В этом случае сера, содержавшаяся в сырье, распределяется между продуктами процесса. При гидрогенизационных процессах происходит более глубокое разрушение сернистых соединений: большая часть их превращается в сероводород и концентрируется в газе.
В табл. 44 представлены примерный выход сухого газа и содержание сероводорода в нем при основных деструктивных процессах переработки нефтяного сырья. Видно, что только одна установка гидрокрекинга мазута высокосернистой нефти мощностью 1 млн. т в год. даст от 2200 до 7700 т сероводорода в год.
Сероводород, получаемый с технологических установок, обычно используют на НПЗ для производства серы, а иногда — для производства серной кислоты. Наиболее распространенным промышленным методом получения серы на основе заводских и природных газов является процесс Клауса, осуществляемый в две ступени327: 2H2S -f 302 ч—^ 2S02 + 2НаО 2H2S + S02 ч=* 2/xSx + 2НаО
Первая реакция идет без катализатора — сероводород сжигают при недостатке воздуха (во избежание дальнейшего окисления S02 в S03). Объем воздуха, поступающего в зону горения, должен dbiTb строго дозирован, чтобы обеспечить для второй ступени процесса требуемое соотношение S02 и H2S. Температура в печи для сжигания H2S в зависимости от концентрации H2S и углеводородов в газе составляет 1100—1300 °С. Печь обычно представляет собой цилиндрический горизонтальный аппарат. Так, на установке, рассчитанной на получение л; 145 т серы в сутки328, печь-реактор имела диаметр 3,66 м и длину 10,7 м. Горелки для газа смонтированы по длине печи или на одном из торцов. По оси печи размещена горизонтальная решетчатая перевальная стенка для лучшего перемешивания газа.
Образование серы начинается уже в первом реакторе. Реакция второй ступени протекает над катализатором — оксидом алюминия. Схема установки представлена на рис. 113. Горячие газы из печи-реактора 1 проходят котел-утилизатор 3, где они охлаждают-
Таблица 44. Выход газа и содержание сероводорода в нем при основных деструктивных процессах переработки сернистых и высокосернистых нефтей
Данные Я. Г. Соркина
| Процесс |
Выход газа на сырье, % (масс.) |
Содержание H2S в газе, % (масс.) |
| Термический крекинг 40%-ного остатка сернистой |
4,0—7,5 | 4,5 |
|
нефти | 29,5 |
|
| Каталитический крекинг вакуумного газойля высо | 6—9 | |
| косернистой нефти |
7,5 | 5,0 ' |
| Замедленное коксование 28%-ного крекинг-остатка | ||
|
смеси восточных нефтей | 9,6 | |
|
Гидроочистка дизельного тойлива из сернистой | 2,5 | |
|
нефти | 11,0 |
|
| Гидрокрекинг мазута .высокосернистой нефти | 2—7 . | |
| Термоконтактный крекинг гудрона высокосернистой | 9—15 | 15,0 |
| арланской нефти |
Примечание. Выход газа и содержание HaS отнесены к «сухому газу*, т. е, «сключая отгон стабилизации. '
ся примерно до 450 °С, чтобы сера оставалась в газовой фазе (температура конденсации паров серы «300°С). Далее газ дополни-
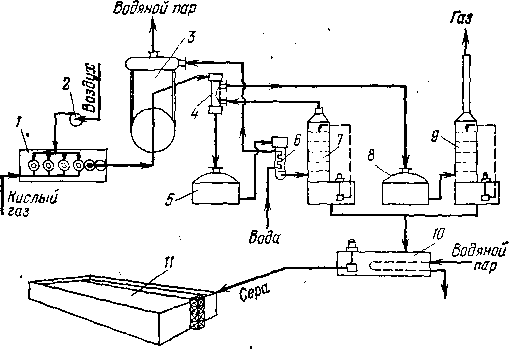
Рис. 113. Схема производства серы (процесс Клауса):
I — печь-реактор; 2 — воздуходувка; 3 — котел-утилизатор; 4 — подогреватель кислого газа; $, 8 — реакторы с катализатором; 6 — экономайзер; 7, 0 —скрубберы; /0 — сборник серы; it — емкость товарной серы.
тельно охлаждают в теплообменнике 4 (не ниже, чем до 340 °С), и газовая смесь поступает в реактор 5, содержащий катализатор.
Взаимодействию Нг5 и S02 благоприятствуют пониженные температуры, следовательно, реакция экзотермична. Поэтому каталитическая часть процесса, в свою очередь, разбита на две ступени (реакторы 5 и 8). Температура на входе в реактор 5 около 340 °С, в реактор 8 около 265 °С; подъем температуры в каждом реакторе составляет примерно 40 °С; объемная скорость подачи газа на катализатор ж 850 ч-1.
Горячие газы после реактора 5 проходят водяной экономайзер 6 и скруббер 7 с насадкой, где происходят дальнейшее охлаждение газов и отделение их от сконденсировавшейся серы, которая в виде расплава стекает с низа скруббера в сборник 10. Газы из скруббера 7 вновь подогреваются в подогревателе 4 и аналогичным путем проходят реактор 8 и скруббер 9, откуда жидкую серу сливают также в сборник 10. Оба скруббера орошаются расплавленной серой.
Содержание сероводорода в исходном газе составляет 75—• 90%, остальное — С02 и следы углеводородов. Наличие углеводородов в кислом газе увеличивает расход воздуха. Полнота извлечения серы 92—95%; разработанная система дополнительной очистки остаточного газа позволила повысить степень извлечения до 98—99%.
Получаемая сера имеет высокую степень чистоты. Большую часть ее расходуют для производства серной кислоты. Если для этой цели использовать в качестве исходного сырья не серу, а непосредственно сероводород, это обходится дороже. Кроме того, серу легко транспортировать к местам производства серной кислоты, которые могут не совпадать с районами расположения НПЗ. Серу используют также в резиновой промышленности, медицине» для производства сероуглерода и в других отраслях народного хозяйства.
